
Autores | Contacto
RE Achenbach *, I Calb ** y LJA Lococo ***
* Jefe de Unidad Dermatología. Hospital General de Agudos “Dr I Pirovano”.
** Ex Jefe de Patología. Hospital “Dr P Piñero”.
*** Médica de Planta. Dermatología. Hospital “Dr I Pirovano”.
Monroe 3550 (1430) Ciudad Autónoma de Buenos Aires. Argentina.
E-mail: rachenbach@hotmail.com
No se declara ningún conflicto de interés.
Dirección
Prof. Dr. Ricardo E. Achenbach
Resumen | Palabras Claves
RESUMEN
Efectuamos una revisión de las denominadas atrofias circunscriptas de la piel, con énfasis en la anetodermia y la atrofodermia; ilustramos algunos casos vividos en los últimos años.
PALABRAS CLAVE: Atrofodermia; Anetodermia.
SUMMARY
A review of the localized atrophies of the skin, focusing in the anetodermia and atrophoderma are made. Some of the cases from our Department of Dermatology are pictured.
KEY WORDS: Anetodermia; Atrophoderma.
Artículo | Referencias
Descargar archivo PDF aquí
ANETODERMIA
El vocablo proviene del griego “anetos” que significa “flojo”o “suelto”, que utilizó por primera vez en 1981 Jadassohn, cuando comunicó el caso de una mujer de 23 años de edad, quien desde los 18 padeció lesiones papulosas, eritematosas y deprimidas en los codos; esas lesiones estaban cubiertas con una piel fina y arrugada1. En 1884, Pellizari describió una alteración similar e infrecuente que llamó “eritema urticado y atrofiante”, de inicio inflamatorio y pruriginoso2. En 1904, Alexander publica un caso semejante pero de inicio ampollar3. En el mismo año Jadassohn, Schweninger y Buzzi4 reportaron el caso de una paciente de 29 años de edad con lesiones de ocho años de evolución, que se distinguían por tener áreas de piel “redundante”, blanquecinas, con herniación de la hipodermis, localizadas en la parte superior del tronco y extremidades superiores, sin fenómenos inflamatorios previos ni ampollas. Desde el siglo XIX se han comunicado muchos casos de esta condición, a la que entre otras denominaciones, se la conoce con el desafortunado término de “atrofia maculosa”.
CLÍNICA
Venecie y Winkelmann efectuaron en 1984, una revisión clínico-patológica en 16 pacientes con anetodermia1,5. En todos los casos, las características clínicas se deben a una disminución o pérdida de las fibras elásticas de la dermis reticular. Por lo general, no existen antecedentes genéticos y no es hereditaria aunque existen raros casos de displasia metafisaria, atrofia óptica y anetodermia, síndrome que se hereda en forma autosómica recesiva6. En 1987, Friedman y col comunicaron dos casos familiares7. La relación hombre-mujer es de 1:1 y la edad promedio de aparición de la enfermedad, corresponde a la segunda década de la vida. Las lesiones son redondeadas u ovaladas y su tamaño oscila entre 1 a 10 cm o más, sin tendencia a confluir. El número de elementos es variable y depende de la causa que la origina, pudiendo oscilar entre una única lesión a cientos de ellas. Se manifiestan como áreas de tejido laxo, que al inicio son pápulas o placas cubiertas con piel fina e hipopigmentada y una particular sensación de depresión o hernia del tejido subcutáneo. En general son asintomáticas y en ocasiones, la piel se observa sobreelevada debido a su laxitud. La localización suele ser en el tronco, los brazos y la nuca, pocas veces la cara; es excepcional el compromiso del cuero cabelludo, las mucosas y el palmo-plantar.
Aunque se han descrito asociaciones con anomalías en el globo ocular: cataratas, queratocono, opacidades corneales; en los huesos: calcificaciones, cifoescoliosis, espina bífida; sistema cardiovascular: prolapso de válvula mitral, insuficiencia aórtica, arritmias e infarto de miocardio, todas ellas se refieren a casos aislados y quizá solo sean coincidencias anecdóticas7.
En la anetodermia secundaria los exámenes complementarios de laboratorio suelen ser anormales, como por ejemplo el síndrome de anticuerpos antifosfolípído. En la idiopática, por lo general, los estudios de laboratorio no suelen encontrar modificaciones.
ETIOPATOGENIA
Su causa es desconocida, aunque parece deberse al aumento de la elastolisis. Con base en los hallazgos de microscopía electrónica, algunos autores consideran que la fagocitosis de las fibras elásticas está aumentada por los macrófagos8, lo que sugiere que los polimorfonucleares neutrófilos contienen una elastasa; este mecanismo podría ser secundario, puesto que la inflamación pocas veces se observa en la clínica. Mediante cultivos celulares se ha establecido, que las fibras colágenas permanecen inalteradas en la mayoría de los casos, aunque en algunos de ellos se hallan disociadas y más finas. La anetodermia ocurre, sobre todo, mediante un fenómeno elastolítico, en el que las gelatinasas A y B, dos metaloproteinasas de matriz poseen actividad degradante de la elastina, ejercen un importante papel en la patogenia de esta afección. La primera puede inducirse al tomar contacto con la interleuquina 1β (factor de transformación de crecimiento β1), en contacto con el colágeno tipo I, que producen los fibroblastos o los queratinocitos. La gelatinasa B solo la producen los fibroblastos y su aumento, se debe a la falla de los mecanismos de control de ambas gelatinasas y de las pro-gelatinasas A y B; es probablemente crucial en el desarrollo y mantenimiento de la anetodermia8,9.
Stephansson y col encontraron que la anetodermia con frecuencia, coexiste en pacientes con anticuerpos antifosfolípido positivos lupus eritematoso sistémico, comparado con los pacientes con LES y anticuerpos antifosfolipídicos negativos; ellos postulan mecanismos inflamatorios y no inflamatorios (microtrombosis)10,11. En un estudio se encontró que 15 pacientes de un grupo de 33 LES y anticoagulante lúpico positivo, tuvieron lesiones de anetodermia10.
En pacientes VIH-1 positivos con aumento de anticuerpos anticardiolipinas, también se registra un aumento de lesiones de anetodermia, en comparación con pacientes VIH-1 positivos y anticuerpos anticardiolipinas normales. La concentración del inhibidor del plasminógeno está aumentada en el paciente VIH-1 +; este inhibidor se une fuertemente a la vitronectina, hecho que contribuye a que esta última disminuya en los tejidos. La vitronectina es necesaria para controlar los niveles de proteinasas incluida la elastasa, la que al aumentar, destruye las fibras elásticas12.
Todos estos hechos total o parcialmente comprobados, aunque atractivos, no explican la totalidad de los casos, los que probablemente correspondan a fenómenos diferentes, que confluyen en una misma lesión cutánea (anetodermia).
HISTOPATOLOGÍA
En el período inicial, las lesiones con aspecto clínico eritematoso, inflamatorias, muestran un denso infiltrado linfocitario perivascular, con predominio de linfocitos T (“helper”). Si bien no se observan alteraciones significativas con la coloración de rutina de hematoxilina y eosina, mediante la técnica de orceína para fibras elásticas, se demuestra una acentuada disminución e inclusive, ausencia completa de dichas fibras en la dermis papilar y reticular superior. Esta alteración se limita exclusivamente a la lesión, ya que las fibras elásticas son normales en la dermis vecina5.
También se ha descrito la existencia de macrófagos y células gigantes multinucleadas, que exhiben fenómenos de fagocitosis de fibras elásticas8.
En el período inflamatorio inicial se demostró por inmunofluorescencia directa, depósitos de IgM y C35.
NOMENCLATURA
Es aún confusa. La bibliografía anglosajona la denominó “atrofia maculosa”, lo que sería erróneo dado que no es una mácula ni una atrofia. La lesión elemental es una pápula o una placa; no tendría sentido clasificarlas en inflamatorias y no inflamatorias, dado que no existe correlación entre la edad de aparición, evolución, asociaciones y respuesta terapéutica, entre ellas. Por lo que se refiere al infiltrado inflamatorio, se lo podrá encontrar o no de acuerdo al estadio evolutivo de la lesión. Las anetodermias se clasificaban convencionalmente en “primarias” y “secundarias”, aunque todas son secundarias (al igual que las atrofias). El concepto de “primaria” tiene validez y es aceptable, si por el mismo se entiende que es de causa desconocida o idiopática. Las anetodermias como todas las afecciones de la piel, tienen una(s) causa(s), no surge(n) por “generación espontánea” y de esta manera, los epónimos que resultan de estas erróneas clasificaciones solo poseen valor histórico13. Las enfermedades que la preceden son por lo general: inflamatorias, tumorales o infecciosas. Cuando no se puede determinar la causa que dio origen a la anetodermia, es válido el concepto de idiopática o primaria14. Las causas más frecuentes de anetodermia secundaria se resumen en el Cuadro I.
CUADRO I
CAUSAS DE ANETODERMIA SECUNDARIA
. Acné (la más frecuente)
. Lupus eritematoso sistémico
. Síndrome de anticuerpos antifosfolípido
. Sífilis
. Sarcoidosis
. Xantomas
. Nevo melanocítico congénito
. Liquen plano
. Neonatos prematuros
. Pilomatrixoma
. Tuberculosis cutánea
. Prurigo nodular
. Picadura de insectos
. Granuloma anular diseminado
. Varicela
. Mastocitosis
. Lepra
. Xantogranuloma
. Moluscos contagiosos
Se comunicó el caso de un paciente con una lepra tuberculoíde polar tratada y curada, con una lesión hanseniana en antebrazo que, al retrogradar y desinfiltrarse dejó como secuela una típica placa de anetodermia, con desaparición de las fibras elásticas, hecho sumamente infrecuente en este tipo de lepra, que es muy común hallarla en la variedad lepromatosa polar15,16.
Los principales diagnósticos diferenciales clínicos de la anetodermia son: la atrofodermia idiopática de Pasini-Pierini, estrías atróficas, atrofia inducida por corticoides y nevo anelástico, aunque el aspecto de las lesiones es característico y de fácil diagnóstico.
La evolución es crónica y la respuesta a los tratamientos ensayados es prácticamente nula. Si las lesiones son pequeñas y escasas pueden ser extirpadas quirúrgicamente. Recientemente se ha comunicado un caso de “atrofia macular en confite”, discutiéndose si se trata de una entidad nueva o no; la clínica es consistente con pequeñas lesiones de atrofodermia pero sin elastolisis histológicamente60.
ELASTOLISIS DE LA DERMIS MEDIA O RETICULAR ALTA
Esta afección fue descrita por Shelley y Wood en 197717, como “arrugas debidas a la pérdida del tejido elástico de la dermis media”, en una mujer sana de 42 años con lesiones urticarianas que, posteriormente, manifestó extensas áreas nacaradas, arrugadas o plegadas finamente, siguiendo las líneas de tensión de la piel. Predomina en mujeres 8 a 1 en la cuarta década de la vida. El aspecto clínico es de piel “envejecida” y al palpar las lesiones, simulan papel de cigarrillo que se pliega fácilmente; pueden adquirir gran tamaño (napas) y una vez constituidas, son asintomáticas. Se localizan en forma simétrica predominantemente, en el tronco y los brazos (áreas expuestas) aunque pueden hacerlo en el cuello, los antebrazos y los glúteos; el compromiso facial es excepcional18,19. Los bordes son netos y observadas con luz tangencial, es posible observar una ligera herniación. En ocasiones se asocia a lesiones papulosas pequeñas, blandas, amarillentas, a veces perifoliculares, que tienden a ubicarse en la corredera vertebral, cuello y región preesternal. Pueden o no observarse fenómenos inflamatorios previos, aunque se ha postulado que siempre existen, siendo en realidad, subclínicos. Carecen de antecedentes familiares, asociaciones o compromiso sistémico. Se han postulado como factores etiopatogénicos la radiación ultravioleta (aunque pueden localizarse en sitios no expuestos) o inmunológicos, por tratamientos anticonceptivos con progesterona o exacerbarse con el embarazo. Las lesiones tienden a progresar lentamente en forma centrífuga, se alivian de manera espontánea y su curso evolutivo es crónico con resistencia a la terapéutica20,21,22,23. En los estadios iniciales la observación microscópica muestra: edema y leve infiltrado linfocitario perivascular. La coloración de orceína para fibras elásticas, demuestra ausencia de dichas fibras que se limitan a la dermis media17. Las fibras elásticas se conservan en la dermis papilar y reticular inferior, al igual que el tejido conectivo perifolicular piloso20,24. En algunos casos se han descrito fagocitosis de las fibras elásticas18. Esta afección no debe confundirse con la elastolisis folicular, debida a una elastasa producida por el Estafilococo epidermidis, que presenta lesiones papulosas pequeñas en el tronco con fenómeno de herniación del ostium folicular24.
El tratamiento con d-penicilamina para la enfermedad de Wilson, puede ocasionar lesiones similares a la elastolisis de la dermis media.
Para algunos autores con un criterio unicista, la elastolisis de la dermis media, elastolisis de la dermis papilar, la elastolisis de la dermis alta simulando un pseudoxantoma elástico y la elastolisis perifolicular, son solo variantes morfológicas de la anetodermia25.
ESTRÍAS ATRÓFICAS
Se conocen como estrías del embarazo o “vergétures”, en comparación con las señales que dejan los latigazos en la piel. Son lesiones anetodérmicas lineales, en forma de huso u onduladas, a cuyo nivel la piel es rosada, violácea o nacarada, delgada, fina, plegada en sectores, blanda y depresible al tacto. Pueden ser numerosas y simétricas; preferentemente se localizan en el abdomen, las mamas, los hombros, la raíz de los miembros, el dorso y los glúteos. Evolucionan en dos etapas: la primera o inflamatoria, en la que se las observa incluso elevadas, en ocasiones eritematosas y pruriginosas y una etapa tardía o cicatrizal, con la piel afinada, fláccida y marfilina-brillante. La causa más común es el embarazo (75-90%), también son comunes durante la pubertad. Los mecanismos de producción serían mecánicos (distensión), endocrinológicos (síndrome de Cushing) y bioquímicos. Otros factores son el uso de corticoides en forma prolongada, desnutrición, infecciones (tuberculosis) y el uso indebido de aparatos para el desarrollo muscular sin control médico, sobre un terreno de predisposición genética que provoca la ruptura de las fibras elásticas26. Recientemente, se comunicaron algunos casos de estrías atróficas27 en pacientes con hipotiroidismo grave, al igual que la aparición de estrías ulceradas secundarias al tratamiento con etretinato28.
La observación histológica muestra, inicialmente, vasodilatación, edema e infiltrado de linfocitos perivasculares en la dermis superficial. La epidermis está adelgazada con desaparición de las crestas interpapilares. Existe una disminución del espesor dérmico. Las fibras colágenas son delgadas con disposición paralela a la superficie cutánea. Las fibras elásticas están engrosadas y se observa aumento relativo de las mismas por estar agrupadas.
El tratamiento, siempre paliativo, puede intentarse con tretinoína tópica, cremas con alfa hidroxiácidos, láser pulsado, microdermoabrasión o en casos especiales, intervención quirúrgica29.
CUTIS LAXA ADQUIRIDO
Es una rara enfermedad que puede ser hereditaria o adquirida, se distingue por piel péndula y pérdida o disminución, con fragmentación de las fibras elásticas de toda la dermis. Las formas adquiridas son infrecuentes y pueden ser generalizadas o localizadas, apareciendo luego de un proceso inflamatorio como: eritema polimorfo, urticaria, síndrome de Sweet o también tumoral como en los mielomas y linfomas. Se han descrito casos en el lupus eritematoso sistémico y el síndrome nefrótico. Las generalizadas predominan en adultos con enfermedad cardiovascular, enfisema o divertículos intestinales. Existe una forma distintiva (Marshall) en niños de Sudáfrica y EEUU, con inflamación intensa y subsiguiente elastólisis30. Las formas localizadas no suelen asociarse con compromiso sistémico, se han descrito en la neurofibromatosis1, sífilis, necrobiosis lipoídica y en los lóbulos auriculares. Recientemente se han comunicado casos de localización acral, post-inflamatoria (urticariana). No existe tratamiento efectivo para el cutis laxa31.
NEVO ANELÁSTICO
Se presenta clínicamente en forma de pápulas planas, rosadas o eritematosas, perifoliculares, asimétricas, localizadas preferentemente en el sector superior del tronco. Existen casos congénitos y adquiridos. La histología muestra pérdida del tejido elástico, con fragmentación de las fibras elásticas en la dermis media y alta. El colágeno no se halla alterado. No se asocia a compromiso sistémico y carece de tratamiento efectivo32.
ELASTORREXIS PAPULOSA
Son pápulas firmes, no foliculares, de 1 a 5 mm de diámetro, blanquecinas, bien delimitadas y distribuidas en el tronco. El examen histológico muestra una fragmentación importante, con una disminución casi completa del tejido elástico de la dermis reticular. Se observa también un infiltrado linfohistiocitario perivascular en la dermis superficial y profunda. No se asocia compromiso sistémico33,34.
PAPULOSIS FIBROSA BLANCA DE LA NUCA
Predomina en adultos de edad media o añosa, de sexo masculino. Las manifestaciones clínicas consisten en pápulas redondeadas u ovaladas, de 2 a 3 mm de diámetro, asintomáticas, múltiples, simétricas que se localizan en la nuca. El estudio histopatológico muestra, en ocasiones, una disminución de la cantidad de melanina en la epidermis. En la dermis papilar y media, coexisten áreas de tejido colágeno engrosado y pérdida del tejido elástico. No hay compromiso sistémico35.
ELASTOLISIS DE LA DERMIS MEDIA SÍMIL PSEUDOXANTOMA ELÁSTICO
Entidad adquirida, puede presentarse con múltiples pápulas asintomáticas o pruriginosas, amarillentas o del color de la piel normal, pequeñas, no foliculares, en “adoquinado”, que comprometen especialmente el cuello. Por coalescencia, pueden formarse placas extensas en el área supraclavicular y pliegues antecubitales y abdominales o inframamarios. No se asocia a las alteraciones sistémicas del que presenta el pseudoxantoma elástico33.
ATROFIA
Desde el punto de vista clínico es la disminución del espesor, consistencia y elasticidad de la piel y forma parte de las lesiones elementales secundarias. Literalmente del latín “atrophía”: falta de desarrollo de cualquier parte del cuerpo, disminución del tamaño, número o ambas cosas a la vez, de uno o varios tejidos de los que forman un órgano, con la consiguiente minoración del volumen, peso y actividad funcional, a causa de la escasez o retardo en el proceso nutritivo; degenerativa: la que va acompañada de un proceso destructor de las células de un tejido; fisiológica: la de algunos tejidos u órganos que en la evolución natural del organismo resultan innecesarios; senil: la de los tejidos y órganos cuando el individuo llega a edad avanzada (Diccionario de la Real Academia Española). A la observación histológica se correlaciona con alteraciones en las fibras colágenas o elásticas de la dermis papilar, reticular, adventicial o incluso del tejido celular subcutáneo y en ciertas afecciones de la epidermis, también puede estar afectada como en el lupus eritematoso discoide crónico.
Siempre es recomendable la maniobra de palpación mediante la “pinza digital”, comparando con la piel aparentemente sana para detectar cambios sutiles, lo mismo que la biopsia contralateral para comparar histológicamente la lesión, con la piel sin alteraciones clínicas.
En este capítulo no se incluyen las numerosas alteraciones genéticas o hereditarias, que se acompañan de atrofia.
ATROFODERMIA IDIOPÁTICA DE PASINI-PIERINI
La describió Pasini en 1923, en una joven de 21 años de edad afectada de tuberculosis y con lesiones cutáneas; la denominó “atrofodermia idiopática progresiva”36. Posteriormente la escuela dermatológica argentina encabezada por Pierini y Borda, publicaron varios casos y eliminaron el término de “progresiva”, al observar que los elementos tienden a permanecer estables, diferenciando una forma “pura”o primaria y otra, que evoluciona a una esclerodermia localizada (morfea)36,37.
Desde el punto de vista clínico comienza con máculas (placas oscuras, únicas o múltiples) redondeadas u ovales, levemente deprimidas respecto de la piel circundante, de tamaño variable entre 2 a varios cm, que pueden confluir sin perder su identificación individual. Los bordes o límites son netos, a menudo se describen como “en pendiente”, son lesiones simétricas, bilaterales y por lo general asintomáticas. La piel que recubre las áreas afectadas es lisa y blanda, aunque pueden existir zonas induradas de morfea y de liquen escleroso y atrófico (atrofoesclerodermia), tal como se ha descrito en la placa de un mismo paciente39. Como lo expresó Abulafia y también otros autores, inicialmente el patrón histológico de esta enfermedad es a veces indistinguible de una morfea39,40. La localización preponderante es en el tronco, aunque pueden observarse también en el abdomen, en los miembros superiores, inferiores y en los hombros. Se han publicado varios casos de topografía sistematizada o zosteriforme41,42. A mayor antigüedad de la lesión, mayor será la pigmentación. Se ha publicado un caso de evolución a una esclerodermia sistémica43, aunque es infrecuente su asociación con alteraciones internas. La etiología es desconocida y el papel de la Borrelia burgdorferi no es en absoluto concluyente44.
Existe controversia respecto de la ubicación nosológica de la atrofodermia idiomática. Para algunos autores constituye una enfermedad independiente, dado que no posee el halo liláceo de la morfea y en general son poco o nada induradas 45. Otros sostienen el concepto que la atrofodermia idiomática, sería una variante morfológica de la morfea de larga evolución. Las principales razones que sustentan este último concepto, son los pacientes con elementos de atrofodermia que llegan a padecer lesiones típicas de morfea, en otras áreas del cuerpo y también de liquen escleroso y atrófico. Las lesiones en el centro de la atrofodermia suelen indurarse y son indistinguibles histológicamente de la morfea (en 1961, 11 de los 22 pacientes descritos y publicados por Pierini, inicialmente habían desarrollado lesiones de morfea46).
Desde el punto de vista histológico conviene realizar dos biopsias: una de la lesión y otra de la piel sana contralateral, para efectuar un estudio comparativo de ambas. La epidermis es normal y puede observarse hiperpigmentación melánica de la capa basal. En los estadios iniciales puede apreciarse vasodilatación, con discretos linfocitos perivasculares y aislados melanófagos en la dermis superficial. No existe alteración de las fibras elásticas, aunque puede haber un aumento relativo de las mismas por estar agrupadas, debido a la disminución del espesor dérmico. Los anexos cutáneos y la hipodermis no se alteran. El hallazgo más significativo al comparar ambas biopsias, es la acentuada disminución del espesor de la piel lesionada comparada con la piel normal39,40,41. Las fibras colágenas pueden engrosarse discretamente en el estadio final44.
La evolución es crónica, aunque algunas lesiones pueden mejorar, excepto la hiperpigmentación. El tratamiento es ineficaz. No consideramos que sea una variante “abortiva” de la morfea sino un estadio final del proceso, aunque aún sabiendo que es una variedad de morfea, creemos que puede mantenerse el epónimo dado lo característico de su clínica.
ATROFODERMIA LINEAR DE MOULIN
En 1992, Moulin describe cinco pacientes con atrofodermia linear, cuyas lesiones se localizaban siguiendo las líneas de Blaschko. Comienza durante la adolescencia sin inflamación previa aparente y permanece estable. Se la considera un mosaicismo debido a una mutación post-zigótica. Happle afirma que los casos de atrofodermia linear o zosteriforme, son en realidad, esta entidad47,48.
ACRODERMATITIS CRÓNICA ATROFIANTE
La describieron e ilustraron Herxheimer y Hartmann en 1902. Constituye una manifestación tardía de la enfermedad de Lyme (Connecticut, EEUU), producida por la picadura de un vector (garrapata del género Ixodes) que transmite la Borrelia burgdorferi y la Borrelia afzeli; esta última es el agente causal de la acrodermatitis crónica atrofiante (ACA). Esta enfermedad es infrecuente en los Estados Unidos, predomina en el norte de Europa, aunque puede aparecer también en otras áreas geográficas. Se produce luego de décadas de la picadura, probablemente por la secreción de citoquinas pro-inflamatorias, aunque no de interferón gama, lo que explicaría su menor capacidad para controlar la infección a diferencia del eritema crónico migrans.
La manifestación clínica es bifásica, en la fase temprana suele ser inespecífica y de difícil diagnóstico, aunque responde favorablemente al tratamiento; en la etapa tardía (la mayoría de los casos) la clínica es típica pero resiste al tratamiento. Su comienzo es insidioso, con cambio de coloración de la piel del pie, talón o dorso de las manos, eritematoso, en ocasiones con un tinte violáceo que puede pasar inadvertido y afectar solo una pierna, difuso o no, con un edema “pastoso” y extensión centrífuga; a veces se extiende al tronco y a la cara. El período inflamatorio persiste durante semanas o meses. Enseguida se inicia con piel lisa, adelgazada (“en papel de cigarrillo”), que facilita la visualización de los vasos del tejido subcutáneo, desaparición de los pelos con disminución o ausencia de secreción sebácea y la sudoración. En algunos casos se producen lesiones hipopigmentadas, que recuerdan a la morfea en su variedad liquen escleroso y atrófico, mientras que en otros casos las lesiones se hiperpigmentan o adquieren aspecto poiquilodermatoso. Es frecuente la extensión del proceso inflamatorio a la cápsula articular y nódulos o placas fibrosas, preferentemente de localización periarticular (rodillas), que dificultan la motilidad; es rara la aparición de úlceras crónicas y calcificaciones. Se puede originar un carcinoma espinocelular en la piel atrófica.
Pueden asociarse otras manifestaciones, como artritis, polineuritis y en la piel, elementos indistinguibles de morfea o liquen escleroso y atrófico.
La histopatología permite apreciar, inicialmente, hiperqueratosis y atrofia epidérmica. La dermis puede ser edematosa y observarse un infiltrado linfocitario en banda, separado por una zona de colágeno normal. El infiltrado linfocitario puede ser perivascular y perianexial40. Existe vacuolización de la capa basal epidérmica con linfocitos, en contacto con la misma y borramiento de la interfase. La unidad infundíbulo-ítsmico-sebácea desaparece con persistencia de las glándulas sudoríparas, que aparentan estar próximas a la epidermis. También disminuye el espesor de la dermis con desaparición gradual de las fibras elásticas50.
El diagnóstico es anamnésico (picadura), clínico, histopatológico y sexológico, con la demostración del germen en las lesiones ya sea por cultivo o PCR. La serología, en general, es positiva con concentraciones elevadas de IgG específica.
El tratamiento recomendado es con ceftriaxona, administrada por vía intravenosa en dosis de 2 gr por día durante 21 días, con buena respuesta si el tratamiento es temprano, aunque incluso casos avanzados pueden reaccionar aceptablemente49,51.
ATROFODERMIA VERMICULATA
Es una alteración relativamente infrecuente, que describió Unna en 1894 (uleritema acneiforme) y que se inicia en la niñez. Entre las múltiples sinonimias se encuentran: “acné vermiculata” (Thibierge, 1900), “atrofodermia reticulada facial” (Pernet, 1916), “foliculitis uleritematosa reticulada” (Mac Kee, 1918). Se hereda en forma autosómica dominante, pero la mayoría de los casos son espontáneos. Se han comunicado casos congénitos y de comienzo en la pubertad o edad adulta, aunque son raros.
Comienza con pequeñas pápulas castaño-pálidas de 1 a 2 mm, que con el transcurso de los meses se transforman en las lesiones típicas. Estas se caracterizan por unas diminutas áreas atróficas, deprimidas (“pits”), simétricas, bilaterales (aunque hay casos unilaterales), reticulares en forma de panal de abeja (“honeycomb atrophy”), localizadas principalmente en las mejillas, con rara extensión de las mismas a labio superior, frente, hélix y lóbulos auriculares. Es frecuente la asociación con queratosis folicular de miembros. Otros elementos que pueden coexistir son: leuconiquia, quistes de milium, tapones córneos prominentes y eritema acompañado de telangiectasias dispuestas en forma reticular. No se observan pápulas o pústulas de acné, seborrea o descamación. La afectación hombre-mujer es uno a uno. En general, no esta acompañada de otras anomalías congénitas o sistémicas, aunque puede ser parte de síndromes de Rombo y aparecer lesiones similares en el cloracné.
La atrofodermia vermiculada pertenece al grupo de las queratosis pilares, con atrofia de la piel estando relacionada al uleritema ofriógeno, también autosómico dominante aunque este último comienza y predomina, por lo general, en las cejas y pestañas. La evolución es crónica, algunos casos pueden involucionan parcialmente, en especial el eritema y los tapones foliculares. La alteración estética es muy intensa con afectación de la calidad de vida del paciente.
En la histopatología suele observarse hiperqueratosis del ostium folicular con atrofia del anexo piloso o bien, folículos pilosos rudimentarios y discreta fibrosis perifolicular. Existe dilatación quística infundibular con comedones y tapones córneos. Las glándulas sebáceas son pequeñas. Puede haber vesículas subcórneas y edema del acrosiringio con formación de milium53.
La terapéutica es difícil y en reemplazo de la dermoabrasión se puede utilizar el láser pulsado52,53.
La atrofodermia nevoide folicular forma parte del síndrome de Bazex (1966), de herencia dominante con poros foliculares dilatados en las extremidades y carcinomas basocelulares en la cara, que aparecen en la infancia o adolescencia. Pueden agregarse hipohidrosis o anhidrosis localizada, con hipotricosis congénita. Por lo que se refiere a atrofodermia folicular, predomina en el dorso de las manos, sector inferior del dorso y superficie extensora de los codos. Se hereda en forma autosómica dominante o ligada al X54.
ELASTOLISIS DE LA DERMIS SUPERFICIAL
Es infrecuente y se caracteriza por presentar pequeñas pápulas, con ausencia de fibras elásticas en la dermis papilar. La causa es desconocida. Desde el punto de vista clínico las lesiones consisten en numerosas pápulas, con un tamaño que oscila entre 0.2 y 0.5 cm y placas amarillentas, cuya aparición puede estar precedida de un prurito discreto. Las lesiones se ubican en el cuello, los hombros, la parte superior del tronco y en general afecta a mujeres mayores55,56,57. La observación histológica demuestra ausencia completa de fibras elásticas en la dermis papilar; su cantidad es normal en la dermis superficial y media. No se aprecian fragmentación ni calcificación de las mismas. Por ultraestructura se demostró la existencia de filamentos rectos, semejantes al amiloide en las fibras elásticas alteradas55,56,57. No existe tratamiento satisfactorio a la fecha.
PANATROFIA DE GOWERS
Es un padecimiento poco frecuente que describió Gowers en 1903 y que se distingue por atrofia localizada, que consiste en la pérdida total o parcial del tejido celular subcutáneo con la atrofia consiguiente.
Esta enfermedad de causa desconocida, afecta preferentemente a mujeres entre los 20 y 50 años de edad; no tiene relación con la esclerodermia.58
Desde el punto de vista clínico, la mayor parte de las lesiones se localizan en la región glútea, el dorso, los brazos y los muslos, aunque también se han descrito algunos casos en los antebrazos y las piernas. En un período de algunas semanas y sin antecedente de proceso inflamatorio cutáneo previo, aparecen depresiones bien delimitadas, con frecuencia simétricas, únicas o en cantidad de dos o tres, de forma oval o redondeada, a veces triangular o cuadrangular que varían en tamaño entre 2 y 20 cm. Al desaparecer en las áreas afectadas, en el lapso de algunos meses la piel suprayacente se atrofia, aunque su aspecto sea normal y las lesiones persisten sin modificación.58
ATROFIA INDUCIDA POR CORTICOIDES
Por lo general es consecuencia de la aplicación inapropiada (contexto clínico erróneo) o por exceso de ésta. No solo se observa por tratamiento tópico (sobre todo los corticoides potentes o bajo oclusión), sino por infiltraciones intradérmicas que alcanzan el subcutáneo o intraarticulares, al retirar la aguja en el recorrido hacia la superficie de la piel, eliminando el corticoesteroide. Los corticoides que se utilizan a través de inhalación también pueden ocasionar atrofia. Los efectos adversos locales y sistémicos inducidos por corticoides se señalan en el Cuadro II.
CUADRO II
LOCALES
– Atrofia dermoepidérmica a veces tan pronunciada que ocasiona fenómeno de herniación o ulceración
– Pseudocicatrices (ancianos)
– Estrías atróficas
– Púrpura
– Telangiectasias
– Fisuras
– Milium
– Rubicundez facial y rosácea por corticoides
– Pseudoacné
– Dermatitis perioral
– Granuloma glúteo
– Hipertricosis
– Hipopigmentación
– Prurito
– Dermatitis por contacto
– Taquifilaxia y fenómeno de rebote
Todos ellos se verán con mayor frecuencia si se utiliza el corticoesteroide en zonas de piel fina, en mucosas, en niños o ancianos, con oclusión o si la barrera córnea se encuentra disminuida.
SISTÉMICOS
– Alteración del eje hipotálamo-hipófisis-suprarrenal
– Glaucoma y catarata subcapsular posterior
– Osteoporosis
– Síndrome de Cushing
Estos últimos dependen de la superficie de piel afectada, la potencia del corticoesteroide y tiempo de aplicación, especialmente si existe automedicación.59
El tratamiento consiste en suspender el corticoesteroide o disminuir su potencia para evitar el fenómeno de “rebote”, utilizar alternativas en forma de monoterapia o alternando con el corticoide menos potente (en lo posible no fluorado). Entre los sustitutos están los derivados análogos de la vitamina D, tacrolimus, azcomicina y pimecrolimus tópicos. El efecto atrofogénico de los corticoides potentes o superpotentes se ha reducido al agregárseles lactato de amonio; estos deberían prescribirse con una frecuencia no mayor de una vez por semana, en virtud que su efecto terapéutico es el mismo que con el uso diario59.
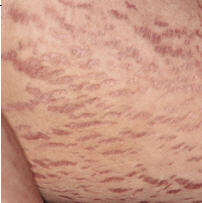
Estrías atróficas (anetodermia)
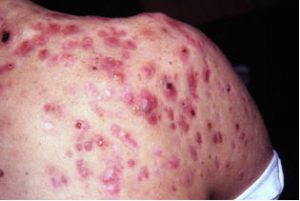
Cicatrices anetodérmicas secundarias a un acné profundo
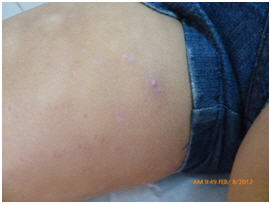
Anetodermia secundaria a moluscos contagiosos (caso reciente, en prensa)
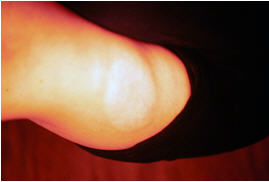
Anetodermia secundaria a lepra tuberculoide (antes y luego de la biopsia de la piel)
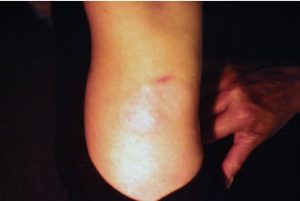
Anetodermia secundaria a lepra tuberculoide (antes y luego de la biopsia de la piel)
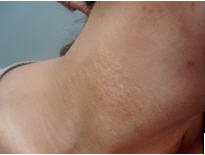
Elastolisis de la dermis media, símil pseudoxantoma elástico
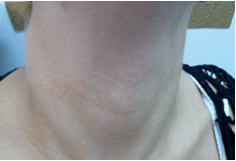
Elastolisis de la dermis media símil pseudoxantoma elástico
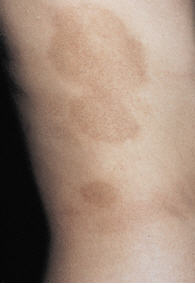
Atrofodermia de Pasini-Pierini
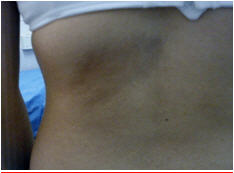
Atrofodermia de Pasini-Pierini
1. Venecie PY, Winkelman RK y Moore BA. Anetoderma: Clinical Findings, Associations, and Long-term Follow-up Evaluations. Arch Dermatol 1984; 120: 1030-1032.
2. Pellizzari C. Eritema orticato atrofizzante: Atrofia parziale idiopatica della pelle. G Ital Mal Ven 1884; 19: 230-243.
3. Alexander A. Mehere Fälle von Hautatrophie. Dermatológica 1904; 11: 338-351.
4. Schweninger E y Buzzi F. Multiple bening tumour-like new growths on the skin. En: Unna PG y col (eds). International Atlas of Rare Skin Diseases. Hamburg. Alemania. Vol 5. Capítulo 15. 1889; 4-5.
5. Venecie PY y Winkelman RK. Histopathological Findings in Anetoderma. Arch Dermatol 1984; 120: 1040-1044.
6. Temtamy SA, El-Meligy M, Badrawyh S y col. Metaphyseal dysplasia, anetoderma and optic atrophy: an autosomal recessive syndrome. En: Bregsma D Editores. Skeletal Dysplasias. Amsterdan. Excerpta Médica 1974; 61-71.
7. Friedman SJ, Venecie PY, Bradley RR y col. Familial anetoderma. J Am Acad Dermatol 1987; 16: 341-345.
8. Zaki I, Scerri L y Nelson H. Primary Anetoderma: Phagocytosis of elastic fibers by macrophages. Clin Exp Dermatol 1994; 19: 338-390.
9. Venecie PY, Bonnefoy A, Gogly B y col. Increased expression of gelatinasas A and B by skin explants in patients with anetoderma. Br J Dermatol 1997; 137: 517-525.
10. Vaalamo M, Kariniemi AL y Shapiro SD. Enhanced Expression of Human Metalloelastasa (MMP-12) in Cutaneous Granulomas and Macrophage Migration. J Invest Dermatol 1999;112 : 499-505.
11. Stephansson EA, Niemi K, Jouhikainen T y col. Lupus Anticoagulant and the Skin. Acta Derm Venereol (Stockh) 1991; 71: 416-422.
12. Stephansson EA y Niemi KM. Antiphospholipid Antibodies and Anetoderma: are they associated?. Dermatology 1995; 191: 204-209.
13. Lindstrom J, Smith K, Skelton H y col. Increased anticardiolipin antibodies associated with the development of anetoderma in HIV-1 disease. Int J Dermatol 1995 ; 34: 408-415.
14. Ackerman AB y col. En: Histologic Diagnosis of Inflamatory Skin Diseases. Williams y Wilkins Editores. Philadelphia. EEUU. 1997; 199.
15. Achenbach RE, Jorge M, Schroh RG y col. Anetodermia secundaria a una lepra tuberculoide polar. Arch Arg Dermatol (en prensa).
16. Magnin PH, Achenbach RE. En: Piense en la Lepra. Talmilep. Alemania. 1990; 30.
17. Shelley WB y Wood MG. Wrinkles due to idiopathic loss of middermal elastic tissue. Br J Dermatol 1977; 97: 441-445.
18. Rae V y Falanga V. Wrinkling due to middermal elastolysis. Arch Dermatol 1989; 125: 950-951.
19. Rudolph RI. Middermal elastolysis. J Am Acad Dermatol 1990; 22: 203-206.
20. Brenner W, Gschnait F, Konrad K y col. Non-inflammatory dermal elastolysis. Br J Dermatol 1978; 99: 335-338.
21. Ortel B, Rappersberg K y Konrad K. Middermal elastolysis in an elderly man with evidence of elastic fiber phagocytosis. Arch Dermatol 1992; 128: 88-90.
22. Marini MA, Saponaro AE y González Cueto DM. Elastólisis de la dermis reticular alta. Rev Arg Dermatol 1995; 76: 148-154.
23. Maghraoui S, Grossin M, Crick B y col. Middermal elastolysis: Report of a case with predominant perifollicular pattern. J Am Acad Dermatol 1992; 26: 490-492.
24. Varadi DD y Saqueton AC. Perifollicular elastolysis. Br J Dermatol 1970; 83: 143-150.
25. Ackerman AB y col. En: A clinical atlas of 101 more common skin diseases with histopathologic correlation. Ardor Scribendi. New Cork. EEUU. 2000; 539.
26. Cordero A (h), Bastué M, y Kien C. Tratamiento de las estrías con una crema a base de ácidos grasos, un estudio a doble ciego. Act Terap Dermatol 1997; 20: 277-280.
27. Niedziela M y Korman E. Atrophic striae. J Pediatr Endocrinol Metab 2001; 14 (7): 901-907.
28. Bowman PH y Hogan DJ. Ulcerated atrophic striae from etretinate. Cutis 2000; 65: 327-328.
29. Mc Daniel D. Laser therapy of strech marks. Dermatol Clin 2002; 20: 67-76.
30. Marshall J, Heyl T y Weber WW. Post-inflammatory elastolysis and cutis laxa. South Afr Med J 1966; 40: 1016-1022.
31. Rongioletti F, Cutolo M, Bondavalli P y col. Acral localized cutis laxa associated with rheumatoid arthritis. J Am Acad Dermatol 2002; 46: 128-130.
32. Staricco, RG y Mehregan HH. Nevus elasticus and nevus elasticus vascularis. Arch Dermatol 1961; 84: 943-947.
33. Lewis KG, Bertcovitch L, Hill SW y col. Acquired disorders of elastic tissue: Part II Decreased elastic tissue. J Am Acad Dermatol 2004; 51: 165-185.
34. Bordas X, Ferrandiz C, Ribera M y col. Papular elastorrhexis: a variety of nevus anelasticus? Arch Dermatol 1987; 123: 433-434.
35. Shimizu H, Nishikawa T y Kimura S. White fibrous papulosis of the neck: a review of 16 cases. Jpn J Dermatol 1985; 95: 1077-1084.
36. Pasini A. Atrofodermia idiopática progresiva. Gior Ital Dermat Sif 1923; 5: 75-79.
37. Pierini LE y Vívoli D. Atrofodermia idiopática (Pasini). Gior Ital Dermat Sif 1936; 77: 403-408.
38. Pierini LE y Bosq P. Atrofodermia idiopática progresiva (Pasini). Rev Arg Derm 1941; 25: 537-539.
39. Borda JM y Abulafia J. Esclerodermia en gotas, atrofodermia idiopática y liquen escleroatrófico coexistentes, alopecia y estado vitiligoide secuelas de esclerodermia. Arch Arg Derm 1956; 1: 98-103.
40. Lever WF y Lever SG. Histopatología de la piel. Sexta edición, Intermédica, Buenos Aires, Argentina, p.435, 1988.
41. Wakelin SH y James MP. Zosteriform atrophoderma of Pasini-Pierini. Clin Exp Dermatol 1995; 29: 244-246.
42. Martínez Fernández M, Miranda-Romero A, Bajo del Pozo C y col. Coexistencia de atrofodermia idiopática de Pasini-Pierini de distribución unilateral con morfea. Med Cut Iber Lat Am 1999; 27: 159-163.
43. Bisaccia EP, Scarbourgh DH, Lowney ED y col. Atrophoderma of Pasini-Pierini and systemic scleroderma. Arch Dermatol 1982; 118: 1.
44. Buechner SA y Rufli T. Atrophoderma of Pasini-Pierini. Clinical and Histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol 1994; 30: 441-446.
45. Cañizares O, Sachs PM, Jaimovich L y col. Idiopathic atrophoderma of Pasini-Pierini. Arch Dermatol 1958; 77: 42-60.
46. Quiroga MI y Woscoff A. L’atrophodermie idiopathique progressive (Pasini-Pierini) et la sclerodrmie atypique lilacee non induree (Gougerot). Ann Derm Syph 1961; 88: 507-509.
47. Moulin G, Hill GV, Barrat D y col. Bandes pigmentées atrophiques accuises suivant les lignes de Blaschko. Am Dermatol Venereol 1992; 119 :729-736.
48. Danarti R, Bittar M, Happle R y col. Linear atrophoderma of Moulin: Postulation of mosaicism for a predisposing gene. J Am Acad Dermatol 2003; 49: 492-498.
49. Fitzpatrick TB y col. En: Dermatology in General Medicine. Tercera Edición Boston. EEUU. Vol I.1993; 1028.
50. Berger BW. Current aspects of Lyme disease and other Borrelia burgdorferi infection. Dermatol Clin 1997; 15 (2): 247-253.
51. Vigliolia PA. Borreliosis. Act Terap Dermatol 2001; 24: 10-29.
52. Handrick C y Alster TS. Laser treatment of atrophoderma vermiculata. J Am Acad Dermatol 2001; 44: 693-695.
53. Frosch PJ, Brumage MR, Prlovic CS y col. Atrophoderma vermiculatum. Case reports and review. J Am Acad Dermatol 1988; 18: 538-542.
54. Novice FM y col. En: Handbook of Genetic Skin Diseases. WB Saunders Company. Philadelphia. EEUU. 1994; 221.
55. El Chariff MA, Masawi AM, Rubeiz NG y col. Pseudoxanthoma elasticum-like dermal elastolysis: a report of two cases. J Cutan Pathol 1994; 21: 252-255.
56. Hashimoto K y Tie MJ. Upper dermal elastolysis. A comparative study with middermal elastolysis. J Cutan Pathol 1994; 21: 533-540.
57. Rongioletti F y Rebora A. Fibroelastolysis patterns of intrinsic skin aging pseudoxanthoma elasticum like papillary dermal elastolysis and white fibrous papulosis of the neck. Dermatology 1995; 191: 19-24.
58. Kluken N y Geir KH. Kasuistischer Beitrag zur Panatrophia cutis localizata. Hautarzt 1965; 16: 422-424.
59. Degreff H y Grossens A. The new corticosteroids: are they effective and safe? Dermatol Clin 1993;11 (1): 155-159.
60. Aksoy B, Ustun H, Gulbahce R y col. Confetti-like macular atrophy: A new entity? J Dermatol 2009; 36: 592-597.
Referencias
REFERENCIAS
1. Venecie PY, Winkelman RK y Moore BA. Anetoderma: Clinical Findings, Associations, and Long-term Follow-up Evaluations. Arch Dermatol 1984; 120: 1030-1032.
2. Pellizzari C. Eritema orticato atrofizzante: Atrofia parziale idiopatica della pelle. G Ital Mal Ven 1884; 19: 230-243.
3. Alexander A. Mehere Fälle von Hautatrophie. Dermatológica 1904; 11: 338-351.
4. Schweninger E y Buzzi F. Multiple bening tumour-like new growths on the skin. En: Unna PG y col (eds). International Atlas of Rare Skin Diseases. Hamburg. Alemania. Vol 5. Capítulo 15. 1889; 4-5.
5. Venecie PY y Winkelman RK. Histopathological Findings in Anetoderma. Arch Dermatol 1984; 120: 1040-1044.
6. Temtamy SA, El-Meligy M, Badrawyh S y col. Metaphyseal dysplasia, anetoderma and optic atrophy: an autosomal recessive syndrome. En: Bregsma D Editores. Skeletal Dysplasias. Amsterdan. Excerpta Médica 1974; 61-71.
7. Friedman SJ, Venecie PY, Bradley RR y col. Familial anetoderma. J Am Acad Dermatol 1987; 16: 341-345.
8. Zaki I, Scerri L y Nelson H. Primary Anetoderma: Phagocytosis of elastic fibers by macrophages. Clin Exp Dermatol 1994; 19: 338-390.
9. Venecie PY, Bonnefoy A, Gogly B y col. Increased expression of gelatinasas A and B by skin explants in patients with anetoderma. Br J Dermatol 1997; 137: 517-525.
10. Vaalamo M, Kariniemi AL y Shapiro SD. Enhanced Expression of Human Metalloelastasa (MMP-12) in Cutaneous Granulomas and Macrophage Migration. J Invest Dermatol 1999;112 : 499-505.
11. Stephansson EA, Niemi K, Jouhikainen T y col. Lupus Anticoagulant and the Skin. Acta Derm Venereol (Stockh) 1991; 71: 416-422.
12. Stephansson EA y Niemi KM. Antiphospholipid Antibodies and Anetoderma: are they associated?. Dermatology 1995; 191: 204-209.
13. Lindstrom J, Smith K, Skelton H y col. Increased anticardiolipin antibodies associated with the development of anetoderma in HIV-1 disease. Int J Dermatol 1995 ; 34: 408-415.
14. Ackerman AB y col. En: Histologic Diagnosis of Inflamatory Skin Diseases. Williams y Wilkins Editores. Philadelphia. EEUU. 1997; 199.
15. Achenbach RE, Jorge M, Schroh RG y col. Anetodermia secundaria a una lepra tuberculoide polar. Arch Arg Dermatol (en prensa).
16. Magnin PH, Achenbach RE. En: Piense en la Lepra. Talmilep. Alemania. 1990; 30.
17. Shelley WB y Wood MG. Wrinkles due to idiopathic loss of middermal elastic tissue. Br J Dermatol 1977; 97: 441-445.
18. Rae V y Falanga V. Wrinkling due to middermal elastolysis. Arch Dermatol 1989; 125: 950-951.
19. Rudolph RI. Middermal elastolysis. J Am Acad Dermatol 1990; 22: 203-206.
20. Brenner W, Gschnait F, Konrad K y col. Non-inflammatory dermal elastolysis. Br J Dermatol 1978; 99: 335-338.
21. Ortel B, Rappersberg K y Konrad K. Middermal elastolysis in an elderly man with evidence of elastic fiber phagocytosis. Arch Dermatol 1992; 128: 88-90.
22. Marini MA, Saponaro AE y González Cueto DM. Elastólisis de la dermis reticular alta. Rev Arg Dermatol 1995; 76: 148-154.
23. Maghraoui S, Grossin M, Crick B y col. Middermal elastolysis: Report of a case with predominant perifollicular pattern. J Am Acad Dermatol 1992; 26: 490-492.
24. Varadi DD y Saqueton AC. Perifollicular elastolysis. Br J Dermatol 1970; 83: 143-150.
25. Ackerman AB y col. En: A clinical atlas of 101 more common skin diseases with histopathologic correlation. Ardor Scribendi. New Cork. EEUU. 2000; 539.
26. Cordero A (h), Bastué M, y Kien C. Tratamiento de las estrías con una crema a base de ácidos grasos, un estudio a doble ciego. Act Terap Dermatol 1997; 20: 277-280.
27. Niedziela M y Korman E. Atrophic striae. J Pediatr Endocrinol Metab 2001; 14 (7): 901-907.
28. Bowman PH y Hogan DJ. Ulcerated atrophic striae from etretinate. Cutis 2000; 65: 327-328.
29. Mc Daniel D. Laser therapy of strech marks. Dermatol Clin 2002; 20: 67-76.
30. Marshall J, Heyl T y Weber WW. Post-inflammatory elastolysis and cutis laxa. South Afr Med J 1966; 40: 1016-1022.
31. Rongioletti F, Cutolo M, Bondavalli P y col. Acral localized cutis laxa associated with rheumatoid arthritis. J Am Acad Dermatol 2002; 46: 128-130.
32. Staricco, RG y Mehregan HH. Nevus elasticus and nevus elasticus vascularis. Arch Dermatol 1961; 84: 943-947.
33. Lewis KG, Bertcovitch L, Hill SW y col. Acquired disorders of elastic tissue: Part II Decreased elastic tissue. J Am Acad Dermatol 2004; 51: 165-185.
34. Bordas X, Ferrandiz C, Ribera M y col. Papular elastorrhexis: a variety of nevus anelasticus? Arch Dermatol 1987; 123: 433-434.
35. Shimizu H, Nishikawa T y Kimura S. White fibrous papulosis of the neck: a review of 16 cases. Jpn J Dermatol 1985; 95: 1077-1084.
36. Pasini A. Atrofodermia idiopática progresiva. Gior Ital Dermat Sif 1923; 5: 75-79.
37. Pierini LE y Vívoli D. Atrofodermia idiopática (Pasini). Gior Ital Dermat Sif 1936; 77: 403-408.
38. Pierini LE y Bosq P. Atrofodermia idiopática progresiva (Pasini). Rev Arg Derm 1941; 25: 537-539.
39. Borda JM y Abulafia J. Esclerodermia en gotas, atrofodermia idiopática y liquen escleroatrófico coexistentes, alopecia y estado vitiligoide secuelas de esclerodermia. Arch Arg Derm 1956; 1: 98-103.
40. Lever WF y Lever SG. Histopatología de la piel. Sexta edición, Intermédica, Buenos Aires, Argentina, p.435, 1988.
41. Wakelin SH y James MP. Zosteriform atrophoderma of Pasini-Pierini. Clin Exp Dermatol 1995; 29: 244-246.
42. Martínez Fernández M, Miranda-Romero A, Bajo del Pozo C y col. Coexistencia de atrofodermia idiopática de Pasini-Pierini de distribución unilateral con morfea. Med Cut Iber Lat Am 1999; 27: 159-163.
43. Bisaccia EP, Scarbourgh DH, Lowney ED y col. Atrophoderma of Pasini-Pierini and systemic scleroderma. Arch Dermatol 1982; 118: 1.
44. Buechner SA y Rufli T. Atrophoderma of Pasini-Pierini. Clinical and Histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol 1994; 30: 441-446.
45. Cañizares O, Sachs PM, Jaimovich L y col. Idiopathic atrophoderma of Pasini-Pierini. Arch Dermatol 1958; 77: 42-60.
46. Quiroga MI y Woscoff A. L’atrophodermie idiopathique progressive (Pasini-Pierini) et la sclerodrmie atypique lilacee non induree (Gougerot). Ann Derm Syph 1961; 88: 507-509.
47. Moulin G, Hill GV, Barrat D y col. Bandes pigmentées atrophiques accuises suivant les lignes de Blaschko. Am Dermatol Venereol 1992; 119 :729-736.
48. Danarti R, Bittar M, Happle R y col. Linear atrophoderma of Moulin: Postulation of mosaicism for a predisposing gene. J Am Acad Dermatol 2003; 49: 492-498.
49. Fitzpatrick TB y col. En: Dermatology in General Medicine. Tercera Edición Boston. EEUU. Vol I.1993; 1028.
50. Berger BW. Current aspects of Lyme disease and other Borrelia burgdorferi infection. Dermatol Clin 1997; 15 (2): 247-253.
51. Vigliolia PA. Borreliosis. Act Terap Dermatol 2001; 24: 10-29.
52. Handrick C y Alster TS. Laser treatment of atrophoderma vermiculata. J Am Acad Dermatol 2001; 44: 693-695.
53. Frosch PJ, Brumage MR, Prlovic CS y col. Atrophoderma vermiculatum. Case reports and review. J Am Acad Dermatol 1988; 18: 538-542.
54. Novice FM y col. En: Handbook of Genetic Skin Diseases. WB Saunders Company. Philadelphia. EEUU. 1994; 221.
55. El Chariff MA, Masawi AM, Rubeiz NG y col. Pseudoxanthoma elasticum-like dermal elastolysis: a report of two cases. J Cutan Pathol 1994; 21: 252-255.
56. Hashimoto K y Tie MJ. Upper dermal elastolysis. A comparative study with middermal elastolysis. J Cutan Pathol 1994; 21: 533-540.
57. Rongioletti F y Rebora A. Fibroelastolysis patterns of intrinsic skin aging pseudoxanthoma elasticum like papillary dermal elastolysis and white fibrous papulosis of the neck. Dermatology 1995; 191: 19-24.
58. Kluken N y Geir KH. Kasuistischer Beitrag zur Panatrophia cutis localizata. Hautarzt 1965; 16: 422-424.
59. Degreff H y Grossens A. The new corticosteroids: are they effective and safe? Dermatol Clin 1993;11 (1): 155-159.
60. Aksoy B, Ustun H, Gulbahce R y col. Confetti-like macular atrophy: A new entity? J Dermatol 2009; 36: 592-597.


