Autores | Contacto
AUTORES
M Mezmezian M *, RE Achenbach **, C Greco ***, A Acevedo ***, L Olvil ****, A Puzzo *****, E Santini-Araujo **** y AJ Lavieri ******
* Médica Patóloga. Hospital General de Agudos Dr. I. Pirovano.
** Jefe de Unidad Dermatología. Hospital General de Agudos Dr. I. Pirovano.
*** Médicos Dermatólogos Concurrentes de Dermatología. Hospital General de Agudos Dr. I. Pirovano.
**** Médicos Patólogos. Práctica Privada.
***** Jefa de Patología. Hospital General de Agudos Dr. I. Pirovano.
****** Médico Dermatólogo de Planta. Hospital General de Agudos Dr. I. Pirovano.
Ricardo E. Achenbach. Hospital General de Agudos “Dr. Ignacio Pirovano”. Av. Monroe 3550 (1430). Ciudad Autónoma de Buenos Aires. Argentina.
E-mail: rachenbach@hotmail.com
No se declaran conflictos de interés.
Recibido: 29.07.2016
Aceptado para su publicación: 31.08.2016
Dirección
Prof. Dr. Ricardo E. Achenbach
Resumen | Palabras Claves
RESUMEN
El mixofibrosarcoma es una neoplasia maligna infrecuente, que puede originarse en los tejidos blandos, se lo ha estadificado como de alto o bajo grado y la localización más frecuente son los miembros inferiores. El reconocimiento clínico del mismo es dificultoso, en primer lugar porque la clínica de otros sarcomas de partes blandas es similar y además semejan lipomas o aún quistes. La extirpación quirúrgica amplia es el tratamiento de elección, por la propensión de este sarcoma a la recidiva local. Los catalogados como de alto grado en un 30 % de los casos, pueden originar metástasis a distancia, especialmente a hueso, pulmón y ganglios linfáticos. La histopatología asegura el diagnóstico en la mayoría de los casos. Comunicamos el caso de un mixofibrosarcoma de bajo grado, en un hombre de 43 años, que a los tres años de control evolutivo, luego de la resección quirúrgica, no presenta recaída local ni distante.
PALABRAS CLAVE: mixofibrosarcoma, neoplasias de tejidos blandos, bajo grado.
SUMMARY
Myxofibrosarcoma is an uncommon soft tissue sarcoma that is grading as low or high malignancy. The principal sites of involvement are the lower limbs. Clinical recognition is difficult since it resembles a cyst, a lipoma, other soft tissue sarcoma or even benign conditions as panniculitis. The wide surgical excision is the main therapeutic approach because local recurrence is frequent. The high grade variant is associated in 30 % of the cases with distant metastasis, especially to the lung, bone and lymph nodes. The histopathological features allow an accurate diagnosis in most cases. A 43 year-old man with a myxofibrosarcoma of the chest wall is reported with a follow-up of two years without recurrence.
KEY WORDS: myxofibrosarcoma, soft tissue tumors, low-grade fibromyxoid sarcoma.
Artículo | Referencias
Descargar archivo PDF aquí
INTRODUCCIÓN
Existen cerca de 12.000 sarcomas en adultos nuevos por año, que comprenden al menos 70 tipos histológicos diferentes, originados en partes blandas, hueso, cartílago, grasa, vasos y tejido conectivo 1. El mixofibrosarcoma (MFS) es un tumor heterogéneo, que tiende a la recidiva local. En una revisión de las historias clínicas de 69 pacientes, 38 hombres con una edad promedio de 62 años, constituyen una de las series más grandes, muestra dificultades en el diagnóstico histológico y por lo general, carecen de fotos clínicas 2,3.
CASO CLÍNICO
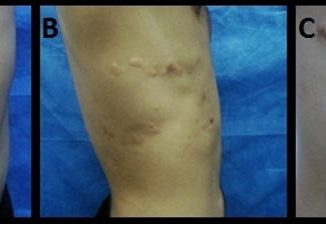
Hombre de 43 años que presentó una masa tumoral, en área infra mamaria de su pared torácica. Dado el carácter indolente y superficial de la lesión, fue derivado al servicio de Dermatología. Al examen físico se observó una masa lisa, depresible de unos 5 x 5 cm, móvil y no adherida a planos profundos, casi asintomática; refería dolor leve solo al ser comprimida (Fig 1).

Fig 1: masa tumoral por debajo de la areola mamaria.
Clínicamente, se sugirió un quiste del tipo pilomatrixoma y se efectuó una resección con escaso margen, diagnóstica y supuestamente curativa. La macroscopía mostró un tumor relativamente bien circunscripto, nodular, de 3 x 3 x 2 cm con un aspecto gelatinoso y compacto. Al examen microscópico, se observó una proliferación multinodular, hipo celular, con una abundante matriz mixoide, escasas y dispersas células “estelares” a fusiformes, núcleos hipercromáticos, pleomórficos y citoplasma eosinofílico. Escasas células vacuoladas o pseudo lipoblastos, se encontraban presentes.
Los vasos eran prominentes, con disposición curvilínea y de paredes delgadas, localizados preferentemente en forma peri tumoral, junto a un infiltrado mixto inflamatorio. No se observaron mitosis atípicas ni necrosis en masa (Fig 2). La inmunomarcación fue positiva para vimentina, negativa para S100 y CD34, el Ki67 era menor al 1%. El diagnóstico final fue de mixofibrosarcoma de bajo grado, por lo que se efectuó una resección completa; el control evolutivo a dos años no mostró recidiva local ni a distancia.

Fig 2: H&E. a- condensación perivascular de las células tumorales; b y c- vasos delicados, curvos, de pared delgada; estroma mixoide; d- núcleos hipercromáticos. a- y b-y; c- 40x y d- 100x.
DISCUSIÓN
Los sarcomas del adulto de partes blandas y de órganos internos, excepto el tumor estromal gastrointestinal (GIST) son infrecuentes, con una incidencia de 4-5/100.000 por año en Europa 4. Los sarcomas de partes blandas cutáneos también son de poca frecuencia; en 10 años de estudio Wollina y col hallaron 67, siendo el más común el fibroxantoma atípico 5.
En un trabajo sobre 192 pacientes con sarcomas recidivantes de la pared torácica, efectuado entre 1989 y 2011, el tipo histológico más frecuente fue el desmoide: 33 pacientes, seguido por el sarcoma pleomórfico indiferenciado: 32 pacientes y el liposarcoma en 22 enfermos. El mixofibrosarcoma se confirmó en 22 también, con la mayor tasa de recurrencias dentro de los tres años de la cirugía 6.
El MFS generalmente se origina en las extremidades de gente añosa; la nomenclatura de estas lesiones ha sido confusa y en 2002, la OMS lo reconoce como entidad propia con características patológicas específicas. Posee una gran tendencia a la recaída local, pero por suerte con escasa capacidad para ocasionar metástasis a distancia, aunque el riesgo de éstas aumenta con cada recidiva 2,7.
Cohortes retrospectivas recientes de MFS, comunicaron que la tasa de recaída local a 5 años de control varía entre 18 a 31 % y la sobrevida total o global es de un 70 %. El tamaño del tumor al momento de la extirpación, los márgenes de resección, necrosis y el número de mitosis se han sugerido como de factores pronósticos, tanto para el tiempo libre de enfermedad como la sobrevida global, aunque los estudios son pequeños en número de pacientes y heterogéneos los tratamientos 2.
La Federación Francesa contra el Cáncer propuso un sistema de gradación, asignando un puntaje para cada categoría dependiendo: del número de mitosis por 10 campos microscópicos de 400x, cantidad de necrosis y de acuerdo con la semejanza o no de la contrapartida celular normal; la gradación final es de 1, 2 ó 3. Existen no obstante, dificultades en otorgar grados a varios sarcomas, entre ellos el alveolar de partes blandas, el de células claras y el epitelioide, entre otros 4.
Los datos cuantitativos por PCR confirman la expresión elevada y consistente de GPRG4 y TNXB, en muestras de MFS a nivel genético; el mismo estudio reveló que los sarcomas de partes blandas, groseramente pueden ubicarse en:
1- sarcoma sinovial,
2- liposarcoma mixoide a células redondas,
3- liposarcoma bien diferenciado con áreas poco diferenciadas y
4- sarcoma pleomorfo a células fusiformes.
De seis tipos histológicos de sarcoma pleomorfo a células fusiformes existen, al menos, entre ellos: liposarcoma desdiferenciado, mixofibrosarcoma, leiomiosarcoma, tumor maligno de la vaina neural periférica, fibrosarcoma y fibrohistiocitoma maligno, todos ellos con expresión genética similar 8.
El MFS comparte algunas de las características histológicas, con los tumores arriba mencionados; además, se puede confundir con patología benigna como: las paniculitis, algunos quistes y la necrobiosis lipoídica. Un caso de recurrencia local de MFS pero de aspecto inflamatorio, ha sido publicado 9.
Van Roggen y col hallaron que hay un subtipo de mixoma celular, que se encontraría entre el mixoma muscular y el mixofibrosarcoma de bajo grado; tal como nuestro caso, tumores como el mixoma juxta-articular, liposarcoma mixoide, MFS de bajo grado, neurofibroma mixoide y el tumor maligno de la vaina neural periférica, son para el diagnóstico diferencial complejo del experto, en sarcomas de partes blandas 10.
El caso aquí presentado posee la ventaja de haber sido extirpado-biopsiado casi parcialmente, lo que permitió luego de efectuado el diagnóstico final, una re extirpación más amplia y un control evolutivo que ya lleva tres años sin recurrencia local, el mayor riesgo del MFS. En general, en los trabajos publicados, no existen o son escasas las imágenes clínicas y la biopsia diagnóstica, se efectúa con punción por aguja, lo que es una clara desventaja 11. Como hecho complementario, el paciente se encuentra bajo tratamiento con terapia sistémica biológica por una psoriasis severa, sin recaída local ni a distancia.
En resumen, el mixofibrosarcoma de bajo grado es un sarcoma de partes blandas, con tendencia a la recaída local y debe ser tenido en cuenta, entre los diagnósticos diferenciales con quistes o supuestos lipomas. Como dermatólogos, necesitamos estar alertas de estos infrecuentes tipos de neoplasias malignas, que parecen indolentes al examen físico. La resonancia magnética nuclear, es un buen complemento para evaluar la extensión de la neoplasia y pensar la estrategia quirúrgica. La cirugía es el tratamiento de elección, con un margen amplio-suficiente para evitar la recaída; si la lesión es mayor a 5 cm la radioterapia puede ser útil 11.
1. Matushansky I, Charytonowicz E, Mills J y col. MFH classification: differentiating undifferentiated pleomorphic sarcoma in the 21th Century. Expert Rev Anticancer Ther 2009; 9 (8): 1135-1142.
2. Hong LJN, Honicek FJ, Raskin KA y col. Prognostic Factors and Outcomes of Patients with Myxofibrosarcoma. Human Pathol 2004; 35 (5). 612-621.
3. Henderson MT, Hollmig T. Malignant fibrous Histiocytoma: Changing perceptions and management challenges. J Am Acad Dermatol 2012; 5: 1-6.
4. Deyrup AT, Weiss SW. Grading of soft tissue sarcomas: the challenge of providing precise information in an imprecise world. Histopathology 2006; 48: 42-50.
5. Wollina U, Koch A, Hansel G y col. A 10-years analysis of cutaneous mesenchymal tumours (sarcomas and related entities) in a skin cancer center. Int J Dermatol 2013; 52: 1189-1197.
6. Mc Millan RR, Sima CS, Moraco NH y col. Recurrence Patterns After Resection of Soft Tissue Sarcomas of the Chest Wall. Ann Thorac Surg 2013; 96: 1223-1228.
7. Kwong R, Kossard S. Histopathological evolution of a cutaneous myxofibrosarcoma. Austral J Dermatol 2008; 49: 169-172.
8. Nakayama R, Nemoto T, Takahashi H y col. Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous Histiocytoma. Modern Pathol 2007; 20: 749-759.
9. Chiu HYI, Chen JS, Hsiao CH y col. Transformation of myxofibrosarcoma into myxoinflammatory fibroblastic sarcoma. J Dermatol 2011; 422-423 (Letter).
10. van Roogen JFG, Mc Menamin ME, Fletcher CDM. Cellular myxoma of soft tissue: a clinic-pathological study of 38 cases confirming indolent clinical behavior. Histopathology 2001; 39: 387-397.
11. The ESMO/European Sarcoma Network Working Group: Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23 (7): 92-99.
Referencias
REFERENCIAS
1. Matushansky I, Charytonowicz E, Mills J y col. MFH classification: differentiating undifferentiated pleomorphic sarcoma in the 21th Century. Expert Rev Anticancer Ther 2009; 9 (8): 1135-1142.
2. Hong LJN, Honicek FJ, Raskin KA y col. Prognostic Factors and Outcomes of Patients with Myxofibrosarcoma. Human Pathol 2004; 35 (5). 612-621.
3. Henderson MT, Hollmig T. Malignant fibrous Histiocytoma: Changing perceptions and management challenges. J Am Acad Dermatol 2012; 5: 1-6.
4. Deyrup AT, Weiss SW. Grading of soft tissue sarcomas: the challenge of providing precise information in an imprecise world. Histopathology 2006; 48: 42-50.
5. Wollina U, Koch A, Hansel G y col. A 10-years analysis of cutaneous mesenchymal tumours (sarcomas and related entities) in a skin cancer center. Int J Dermatol 2013; 52: 1189-1197.
6. Mc Millan RR, Sima CS, Moraco NH y col. Recurrence Patterns After Resection of Soft Tissue Sarcomas of the Chest Wall. Ann Thorac Surg 2013; 96: 1223-1228.
7. Kwong R, Kossard S. Histopathological evolution of a cutaneous myxofibrosarcoma. Austral J Dermatol 2008; 49: 169-172.
8. Nakayama R, Nemoto T, Takahashi H y col. Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous Histiocytoma. Modern Pathol 2007; 20: 749-759.
9. Chiu HYI, Chen JS, Hsiao CH y col. Transformation of myxofibrosarcoma into myxoinflammatory fibroblastic sarcoma. J Dermatol 2011; 422-423 (Letter).
10. van Roogen JFG, Mc Menamin ME, Fletcher CDM. Cellular myxoma of soft tissue: a clinic-pathological study of 38 cases confirming indolent clinical behavior. Histopathology 2001; 39: 387-397.
11. The ESMO/European Sarcoma Network Working Group: Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23 (7): 92-99.