Autores | Contacto
Autores: Soto Andrea,1 Villani Maria Emilia,2 Muñoz Fabio2 y Merola Gladys4
- 1 Médica de planta de Dermatología, Hospital Teodoro Álvarez, Buenos Aires, Argentina
ORCID: 0009-0008-1421-3054 - 2 Médica de planta de Dermatología, Hospital Teodoro Álvarez, Buenos Aires, Argentina
ORCID: 0009-0009-0754-1751 - 3 Médico de planta del servicio de Hemodinamia, Hospital Fernández, Buenos Aires, Argentina
ORCID: 0009-0007-5127-6751 - 4 Jefe de División de Servicio de Dermatología, Hospital Teodoro Álvarez, Buenos Aires, Argentina
ORCID: 0009-0008-6936-3105
Hospital Teodoro Álvarez, Dr. Juan Felipe Aranguren 2701, C1406 CABA
Hospital Fernández, Av. Cerviño 3356, C1425, CABA
Contacto autor responsable : Andrea Soto, andicsoto@gmail.com
- Recibido: 04/01/2023
- Recibido 1°Corrector: 20/08/2023
- Recibido 2° corrector: 03/11/2023
- Aceptado para su Publicación: 7/01/2024
Los autores declaramos no poseer ningún tipo de conflicto de interés
Resumen | Palabras Claves
RESUMEN:
El Síndrome de Parkes Weber se caracteriza por sobrecrecimiento de una extremidad asociado a malformación capilar, venosa, linfática y arteriovenosa. Se presenta el caso de una paciente de 23 años de edad que acudió al Servicio de Dermatología por presentar una úlcera de novo desarrollada sobre malformación capilar congénita. Nos fue posible arribar al diagnóstico utilizando diversos estudios de imágenes, mientras que la medida terapéutica, con el carácter de profilaxis terciaria, consistió en embolizaciones múltiples.
PALABRAS CLAVE: Síndrome de Parkes Weber, cortocircuito arteriovenosos.
ABSTRACT:
Parkes Weber Syndrome is characterized by overgrowth of a limb with capillary, venous, lymphatic, and arteriovenous malformation. We present the case of a 23-year-old patient who came to the dermatology service for an ulcer on congenital capillary malformation. We were able to arrive at the diagnosis using various imaging studies, while the therapeutic measure, with the character of tertiary prophylaxis, consisted of multiple embolizations.
KEY WORDS: Parkes Weber syndrome, arteriovenous shunts.
Artículo
Descargar archivo AQUÍ
INTRODUCCIÓN:
Las malformaciones vasculares son anormalidades de los vasos presentes desde el nacimiento debido a defectos en la angiogénesis durante el desarrollo embrionario. Estas puedan ser de flujo bajo como las capilares, las venosas y las linfáticas, o de flujo alto como las arteriovenosas. En la última modificación, de 2018, de la Clasificación de la ISSVA se incluyen las anomalías vasculares relacionadas con otras anomalías, y dentro de estas se encuentra el Síndrome de Parkes Weber (SWP). Este representa una combinación compleja de malformación capilar, venosa, linfática y malformaciones arteriovenosas.1-2
CASO CLÍNICO
Paciente de sexo femenino de 23 años de edad, oriunda de Bolivia, con antecedentes personales de una malformación vascular congénita en miembro inferior derecho, diagnosticada en su país de origen, sin antecedentes familiares patológicos de importancia. Consultó a nuestro servicio por presentar una úlcera sobre dicha malformación, posterior a un traumatismo local, de seis meses de evolución. La úlcera le producía importante dolor, según escala de dolor EVA cuatro, y ocasional sangrado.
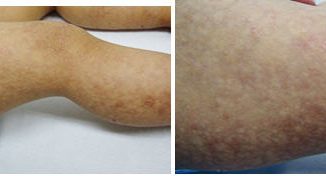
Al examen físico presentaba una mácula color rojo vinoso de contornos netos e irregulares, con una vitropresión negativa, que se iniciaba en la cara posterior de muslo derecho y luego se extendía por la cara lateral externa para alcanzar finalmente la rodilla homolateral. Sobre dicha mácula, y a nivel del tercio inferoexterno del muslo y rodilla derecha, presentaba una úlcera de bordes netos con fondo eritematoso, cubierta por una costra hemática. La piel perilesional se encontraba hiperpigmentada con algunas costras adherentes (Figura 1). Además, comprobamos asimetría de ambos muslos con una diferencia de cuatro cm entre ambos diámetros. A la palpación pudimos constatar un aumento de la temperatura local y la existencia de frémito.
El ecodoppler arteriovenoso de los miembros inferiores mostraba derivaciones arteriovenosas de flujo alto a predomino suprapatelar derecho. Los estudios de laboratorio rutinarios, incluido un coágulograma, fueron normales. La angioresonancia magnética (MRA) evidenció un aumento de la circunferencia del muslo derecho comparada con su homólogo contralateral, a expensas del aumento del espesor del tejido celular subcutáneo de la región posterolateral del muslo. En el seno del mismo se observaron múltiples estructuras vasculares dilatadas tortuosas, de aspecto serpiginoso, y la coexistencia de vasos de flujo lento y flujo rápido. Aquellas se extendían desde el tercio medio del muslo, sector lateral de la rodilla hasta el tercio proximal de la pierna. En la secuencia de MRA realizada se evidenció imagen compatible con malformación arteriovenosa, con llenado precoz de estructuras vasculares venosas, expresando la presencia de un cortocircuito arteriovenoso (Figura 2).

Así, ante una malformación capilar, venosa, arteriovenosa e hipertrofia del miembro afectado se llegó al diagnóstico de Síndrome de Parkes Weber, y se lo pudo ubicar dentro del estadio III según la clasificación de Schobinger R., de la historia natural de las MAV.
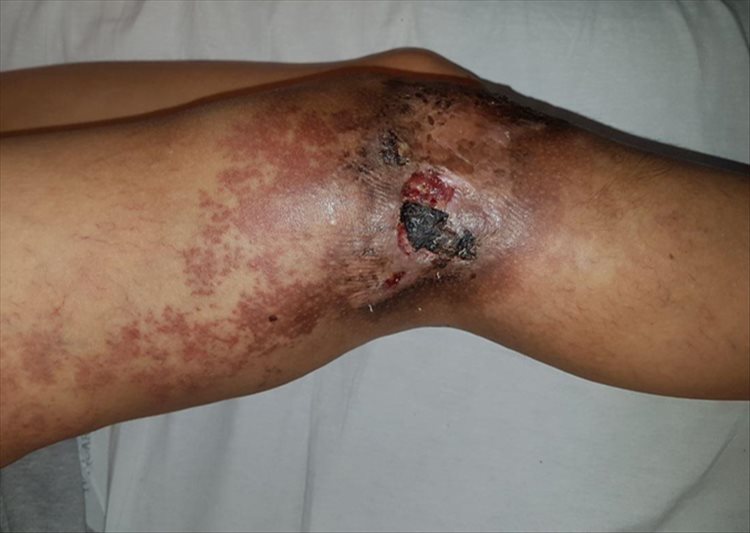
Se realizaron interconsultas con el Servicio de Cardiología, que mediante un ecocardiograma transtorácico descartó insuficiencia cardíaca; sin embargo, se evidenció hipertensión pulmonar de grado leve. El Servicio de Traumatología indicó una goniometría para medición de los huesos largos, que demostró la simetría longitudinal del miembro afectado. Fue evaluada por el Servicio de Cirugía Vascular y Hemodinamia del Hospital Fernández, donde llevaron a cabo una angiografía para determinar extensión y localización exacta de la malformación, y posteriormente realizar un procedimiento endovascular consistente en embolización percutánea. Las mismas fueron realizadas con un adhesivo tisular de cianobutilnacrilato y lipiodol, en tres tiempos, y se observó cierre casi total de la úlcera (Figura 3).

DISCUSIÓN
El síndrome de Parkes Weber es una rara entidad, reconocido por primera vez en el año 1907 por Frederick Parkes Weber. Se caracteriza por una malformación capilar extensa segmentaria localizada en un miembro, acompañado de venas varicosas, hipertrofia del miembro afectado y múltiples malformaciones arteriovenosas (MAV) por debajo de la misma. Estas últimas le dan el sello a la enfermedad.1
Presenta una incidencia <1.5 % de la población general. No se ha encontrado predilección por sexo, raza o ubicación geográfica.3-4 La mayoría de los casos son esporádicos, debido a mutaciones somáticas.5 Sin embargo, algunos son a causa de mutaciones en el gen RASA 1 situado en el brazo largo del cromosoma 5 (5q13.3), con un patrón de herencia autosómica dominante. En estos casos, las personas afectadas, por lo general, tienen múltiples malformaciones capilares.6-8 El primer caso familiar fue descripto en el 2006.9 El gen RASA1 codifica una proteína conocida como p120RASGAP, que está implicada en la transmisión de señales químicas que ayudan al crecimiento y proliferación celular, la diferenciación y el movimiento celular. Se han identificado varias mutaciones en el gen RASA 1 en personas con SPW.1
En general, los pacientes tienen afectación de miembros inferiores, y es menos frecuente el compromiso de los miembros superiores.4 Las lesiones cutáneas están presentes al nacimiento y empeoran con la edad. La malformación capilar suele estar representada por unas máculas color rojo-vinoso, violácea o rosada, planas o a veces levemente sobreelevadas, de aspecto geográfico que no desaparecen a la vitropresión.10-11 La malformación venosa aparece como masas azuladas, frágiles y fácilmente compresibles, no pulsátiles, que generalmente se agrandan con la maniobra de Valsalva. En cuanto a las linfáticas, pueden presentarse como linfedema o lesiones quísticas, siendo estas poco frecuentes.1
El sobrecrecimiento puede estar presente al momento del nacimiento o desarrollarse con los años, asociado con el estasis venoso y linfático y con el crecimiento longitudinal óseo o del diámetro del miembro afectado por el compromiso de las partes blandas.1,4 Fernández de Pineda reportó un caso de SPW con una diferencia entre los diámetros de las extremidades inferiores un poco menor de 12 cm, pero con el acortamiento del fémur del miembro afectado.11
A la palpación se puede constatar aumento de temperatura local, várices pulsátiles y frémito, signos de una malformación arteriovenosa subyacente de flujo alto. Las mismas son lesiones vasculares compuestas por vasos arteriales dismórficos conectados a vasos venosos directamente, sin la intervención del lecho capilar. En la pubertad o tras un traumatismo o un procedimiento quirúrgico, suelen entrar en un período de empeoramiento progresivo.3 (Tabla 1)
El paciente refiere síntomas de la tétrada de Celso (dolor, calor local, rubor, edema), fatiga, incluso el signo de Virchow (impotencia funcional). La evolución fisiopatológica de las MAV se explica por la compresión mecánica local sobre las estructuras circundantes y por el impacto hemodinámico de la derivación arteriovenosa en la circulación.1 Así, las principales complicaciones son insuficiencia cardíaca, hipertensión pulmonar e isquemia cutánea con la formación de úlceras y sangrados, como en el caso de nuestra paciente. Algunos pacientes pueden requerir amputación del miembro afectado. También puede haber tromboflebitis y, en los peores desenlaces, coagulación intravascular diseminada.3,12-15 El riesgo de mortalidad es del 1 %.4
El síndrome de Klipplel-Trenaunay (SKT) debe ser el primer diagnóstico diferencial, ya que estas dos entidades tienen una significativa diferencia tanto en la evolución clínica como en su pronóstico. El SKT es una afección estrictamente de flujo bajo, y son más características las malformaciones venosas y linfáticas. La malformación capilar es de color rojo vinoso y de tono más oscuro en este síndrome, a diferencia del SPW.16
Tabla 1. Etapas de la historia natural de una MAV
| I. Quiescente | Mancha rosa azulada, eritema caliente y derivación arteriovenosa |
| II. Expansión | Estadio I + hipertrofia, pulsaciones, frémito, , soplos y venas tortuosas |
| III. Destrucción | Estadio II + alteraciones cutáneas: distrofia, úlceras, hemorragias, dolor y necrosis |
| IV. Descompensación | Estadio III + insuficiencia cardíaca |
Tomado de Schobinger R. Proceedings of International Society for the Study of Vascular Anomalies Congress Rome, Italy; 23-26 June. 1996
El SPW se diagnostica por signos clínicos y resultados de imágenes. El ecodoppler es el estudio inicial para determinar la existencia de lesiones de flujo alto. La TAC, la RMN y la angiografía son útiles para confirmar la existencia de cortocircuitos arteriovenosos y para determinar la extensión de las lesiones. La angiografía de contraste se considera la prueba de referencia, y generalmente se reserva a los pacientes que son candidatos potenciales para la embolización.1 Tanto los estudios radiológicos como la medición del diámetro de ambas extremidades deberán repetirse periódicamente. Las anormalidades venosas y las linfáticas suelen ser estudiadas con flebografía y linfografía, respectivamente.
Los objetivos de las medidas terapéuticas profilácticas son disminuir la progresión de la enfermedad y las complicaciones, y mejorar la calidad de vida de los pacientes.17-18 Durante la infancia, suele ser conservadoras, con intervenciones paliativas, minimizando los traumatismos. Es útil el uso de medidas elásticas de compresión desde los tres meses de edad.4,1 No se ha encontrado ningún tratamiento farmacológico efectivo. Existen drogas antiangiogénicas, como el bevacizumab y el ranibizumab, que podrían ejercer efecto beneficioso en estos pacientes, pero se precisan estudios que avalen su uso.19 En la edad adulta deben considerarse procedimientos invasivos según las eventuales complicaciones del SPW. La profilaxis terciaria de elección en las MAV es la embolización endovascular. La recomendación es realizar múltiples intervenciones para evitar riesgos como grandes sangrados o necrosis.1,16 El objetivo principal es la oclusión del nido vascular central y no de algunas de la/s rama/s de la malformación arteriovenosa de manera individua. La cirugía es otra opción, pero no aislada sino con la interacción sinérgica de la embolización. Se reportó un caso con colocación de stent en la malformación, con buenos resultados.17
CONCLUSIONES
Hemos arribado a diagnóstico de SPW en una paciente adulta. Por presentar una úlcera en la región distal del muslo y cara externa de la rodilla del miembro inferior derecho, desarrollada sobre una MAV, le fueron efectuados variados estudios de imágenes, gracias a los cuales hemos reunido criterios que nos permiten incluir esta anomalía dentro del estadio III de la clasificación de Schobinger de las etapas de la historia natural de las malformaciones arteriovenosas. Por otro lado, y puesto que la joven enferma cursaba, si bien de forma leve, con hipertensión pulmonar, debieran realizarse evaluaciones periódicas en el Servicio de Cardiología, pues de desarrollar insuficiencia cardíaca implicaría la progresión al estadio IV de la clasificación mencionada. Consideramos de suma importancia el diagnóstico precoz, como buena práctica de profilaxis secundaria y terciaria, la cual resultará en una mejoría de la calidad de vida de estos pacientes. Además, es adecuado un estrecho seguimiento de su evolución durante un tiempo prologado.
Deseamos destacar la buena respuesta a la técnica de la embolización, considerando la alta morbimortalidad descrita para este procedimiento.
Los nuevos descubrimientos en el campo de la genética referidos a su participación en la etiopatogenia de las malformaciones vasculares ofrecen datos que representan una esperanza para el desarrollo de tratamientos prometedores para estos trastornos.
Referencias
REFERENCIAS:
- Banzic L, Brankovic M y Maksimovic Z. Parkes Weber syndrome-Diagnostic and management paradigms: A systematic review. 2017; 32(6): 371-383. PMID: 27511883. DOI: 10.1177/0268355516664212
- Wang MX, Kamel S, Elsayes KM et al. Vascular Anomaly Syndromes in the ISSVA Classification System: Imaging Findings and Role of Interventional Radiology in Management. Radiographics. 2022 Oct; 42(6): 1598-1620. PMID: 36190850 DOI: 1148/rg.210234
- Sierra-Juárez MÁ, Córdova-Quintal P, Fabián-Mijangos W et al. Síndrome de Parkes Weber; Schobinger IV. Rev Mex Angiol 2013; 41(2): 78-81.
- Giron-Vallejo O, Lopez-Gutierrez JC y Fernandez Pineda I. Diagnosis and treatment of Parkes Weber syndrome: a review of 10 consecutive patients. Ann Vasc Surg 2013; 27: 820-825. DOI: 1016/j.avsg.2013.01.001
- Kondapavuluri BK, Bharadwaj RN, Shaikh S et al. Parkes Weber syndrome involving right lower limb: a case report. Indian J Surg 2015; 77: 130-134. PMID: 25972672 IDPM: PMC4425771 DOI: 1007/s12262-014-1201-8
- Revencu N, Boon LM, Mulliken JB et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 2008; 29: 959-965. DOI: 10.1002/humu.20746.
- Brunetti-Pierri N, Seidel GF, Levy ML et al. Parkes Weber syndrome occurring in a family with capillary malformations. Clin Dysmorphol 2007; 16: 167-171. PMID: 17551330 DOI: 1097/MCD.0b013e3280f6cff2
- Eerola I, Boon LM, Mulliken JB et al. Capillary Malformation–Arteriovenous Malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003; 73(6): 1240-1249. PMID: 14639529 PMCID: PMC1180390 DOI: 1086/379793
- Courivaud D, Delerue A, Delerue C, Boon L, Piette F y Modiano P. Familial case of Parkes-Weber syndrome. Ann Dermatol Venereol 2006; 133: 445-447. DOI: https://doi.org/10.1016/s0151-9638(06)70936-x
- Reta Sanchez A, Peniche J y Arellano I. Sindrome de Parkes Weber. Dermat Rev Mex 1996; 40 (4): 275-277.
- Fernandez-Pineda I y Lopez-Gutierrez JC. Parkes Weber syndrome associated with a congenital short femur of the affected limb. Ann Vasc Surg 2009; 23: 257e1-257e2. PMID: 18835518 DOI: 1016/j.avsg.2008.08.021
- Carmona-Ruiz H, Heredia-Gutiérrez JC, Orihuela-Rodríguez O. Pulmonary hypertension associated with Parkes-Weber syndrome (a rare congenital arteriovenous malformation). Vasc Med. 2017 Apr; 22(2): 162-163. PMID: 27707981 DOI: 1177/1358863X16668264
- Picolini A, Carlessi A, Heredia G y Moles V. Insuficiencia cardiaca de alto gasto por fistula arterio-venosa congénita Sindrome de Parkes Weber. Ins Card 2010; (5)3: 150-153.
- Hoffmann F, Sondermann W, Alkhater M, Weindorf N y Dissemond J. Chronic foot ulcer caused by Parkes Weber syndrome. Int Wound J. 2016 Oct; 13(5): 1092-1094. DOI: 1111/iwj.12617
- Patel R, et al. BMJ Case Rep 2021; 14:e241649. doi:10.1136/bcr-2021-241649
- Ziyeh S, Spreer J, Rossler J et al. Parkes Weber or Klippel-Trenaunay syndrome? Non-invasive diagnosis with MR projection angiography. Eur Radiol 2004; 14: 2025-2029. PMID: 15007616 DOI: 1007/s00330-004-2274-8
- Acar Z, Kiris A, Bektas H y Erden T. Short- and mid-term effects of covered stent implantation on extremity findings and heart failure in Parkes Weber syndrome: a case report. Eur Heart J Case Rep. 2020 Mar 10; 4(2): 1-4. PMID: 32352073 IDPM: PMC7180698 DOI: 1093/ehjcr/ytaa046
- He B, Sun MS, Liu JW, Nie QQ, Fan XQ y Liu P. Major Limb Amputation in Parkes-Weber Syndrome With Refractory Ulceration: A Case Report and Literature Review. Int J Low Extrem Wounds. 2023 Mar; 22(1): 168-173. DOI: 10.1177/1534734620986683
- Martinez-Lopez A, Salvador-Rodriguez L, Montero-Vilchez T et al. Vascular malformations syndromes: an update. Curr Opin Pediatr. 2019; 31(6): 747‐ PMID: 31693582 DOI: 10.1097/MOP.0000000000000812