
Autores | Contacto
C. Wolf
Laboratorio de Investigaciones Clínicas. Departamento de Medicina Occidente. Facultad de Medicina. Universidad de Chile. Hospital San Juan de Dios. Santiago de Chile. Chile. Casilla 33052. Correo 9. Teléfono: 681 7414 E-mail: cwf1255@yahoo.es
Fecha de recepción: 12.04.07
Fecha de aceptación: 20.06.07
Dirección
Prof. Dr. Ricardo E. Achenbach
Resumen | Palabras Claves
RESUMEN: La hiperpigmentación cutánea por melanina en zonas expuestas al sol puede estar asociada a un desequilibrio en la homeostasis del hierro. La hepcidina es un péptido responsable de la regulación negativa de la absorción del hierro en el intestino delgado y de su liberación por los macrófagos. Posee capacidad antimicrobiana. Es sintetizada en el hígado, secretada al torrente circulatorio y excretada por la orina. La sobreexpresión causa anemia y su déficit, sobrecarga de hierro (acumulación en diferentes órganos y hemocromatosis hereditaria). Los antagonistas de la hepcidina podrían utilizarse en el tratamiento de la anemia resistente a eritropoyetina, asociada a procesos crónicos. Por su parte, los agonistas o sustancias que estimulen la producción de hepcidina, podrían constituir un tratamiento en enfermedades con sobrecarga de hierro (siderosis) y por consiguiente, corregir la hiperpigmentación asociada.
PALABRAS CLAVE: Hepcidina; Hiperpigmentación cutánea; Metabolismo del hierro; Anemias inflamatorias; Hemocromatosis.
SUMMARY: The cutaneous hyperpigmentation by melanin in zones of the skin exposed to the sun can be associated to an imbalance in the homeostasis of the iron. The hepcidin is a peptide responsible for the negative regulation of the absorption of the iron in the small intestine and of its liberation by the macrophages. It has, in addition, antimicrobial capacity. It is synthesized in the liver, secreted to the circulatory torrent and excreted by the urine. Its overexpression causes anemia and its deficit iron overload (accumulation in different organs and hereditary hemochromatosis), The antagonists of the hepcidin, could be used in the treatment of anemia resistant to erythropoyetin associated to chronic processes. On the other hand, the agonists or substances that stimulate the hepcidin production, could constitute a treatment in diseases with overload of iron (siderosis) and therefore, to correct the associate.hyperpigmentation.
Artículo | Referencias
Descargar archivo PDF aquí
Diversas y heterogéneas enfermedades o situaciones cursan con hiperpigmentación de tipo melánica difusa, entre ellas se destacan trastornos endócrinos como la enfermedad de Addison y tumores de la hipófisis; ingesta de ciertas drogas o metales y diversos trastornos metabólicos como carencias vitamínicas por desnutrición; enfermedades hepáticas crónicas como la porfiria cutánea tarda y la hemocromatosis. Estas dos últimas enfermedades se caracterizan por presentar acumulación excesiva de hierro, haciendo pensar que ambas están relacionadas desde un punto de vista genético 1.
El objetivo de este trabajo es presentar una puesta al día de las proteínas (péptidos) involucradas en la homeostasis del hierro. El hierro (Fe) es considerado un micronutriente por la escasa cantidad que se encuentra en el organismo: 1/23 000 del peso corporal, aun así, si su aporte o disponibilidad disminuye, se produce anemia asociada. Un incremento de hierro lleva a la acumulación en diferentes órganos y produce daño celular por mecanismos que involucran el estrés oxidativo.
Diversas proteínas están relacionadas con la homeostasis del hierro: la transferrina, la ferritina y el receptor de la transferrína. Además de otras, tales como los denominados elementos de respuesta al hierro (IRE: iron responsive elements) y la proteína reguladora del hierro (IRP: iron regulatory protein). El receptor de transferrina 2 (TfR2), el transportador de metales pesados (DMT1 o Nramp2), la ferroportina, la hefaestina, el citocromo b duodenal (citb D), la proteína HFE vinculada a la hemocromatosis hereditaria, la hemojuvelina (proteína que se corresponde al gen HFE2 responsable de la hemocromatosis juvenil).
La hepcidina es una proteína recientemente descrita que tiene un rol central en la homeostasis del hierro.La capacidad del hierro de existir en dos estados de oxidación, lo hace un elemento tóxico. El hierro libre es capaz de generar radicales libres que dañan a lípidos, proteínas, ADN y otros compuestos celulares. De este modo, el exceso de hierro está relacionado con el estrés oxidativo y el envejecimiento.
Estudios recientes, realizados en población adulta mayor, demostraron que sólo el 2,7 % tiene deficiencia de hierro en comparación al 13,0 % que presenta sobrecarga de él 2. En condiciones fisiológicas, la cantidad total de Fe en el organismo es dependiente de su absorción intestinal y en ella intervienen mecanismos regulados genéticamente. En numerosas enfermedades, genéticas o adquiridas, se puede producir acumulación excesiva de este metal, lo que se conoce como hemosiderosis. Se habla de hemocromatosis cuando el exceso de Fe en el organismo se debe a una absorción intestinal desmedida, a consecuencia de una alteración genética y se depositan en diferentes tejidos, con compromiso anatómico y funcional. En general, en estos casos el contenido total de Fe en el organismo es superior a 5 g.
La hemocromatosis hereditaria fue descrita por Troisier 3 a fines del siglo XIX, pero fue von Recklinghausen 4 quien primero se refirió a ella como hemocromatosis. En 1976 se encontró que el gen responsable de la enfermedad estaba ligado al sistema HLA en la rama corta del cromosoma 6 y en 1996 se identificó el gen de la hemocromatosis denominándosele HFE.
Inicialmente se pensaba que ésta era una única entidad, pero en los últimos años se han descrito al menos cinco variedades que tienen características clínicas y genéticas bien definidas (Tabla I).

Se considera a la hemocromatosis como la enfermedad genético – metabólica más frecuente en la población caucásica, especialmente de origen celta, en quienes la mutación asociada a la enfermedad se puede encontrar en 1 de 200 individuos y en alrededor del 10 a 15%, la expresión de la enfermedad 5.
La hemocromatosis o hemosiderosis primaria es una enfermedad idiopática y familiar debida a absorción intestinal aumentada de hierro. Es más frecuente en el hombre que en la mujer en una relación de 9:1. Se expresa, en general, alrededor de los 40 años. Los órganos más afectados por la acumulación de hierro son el hígado, donde produce fibrosis y con el tiempo, cirrosis; el páncreas en el que compromete la producción de insulina derivando en diabetes bronceada, denominada así por el color oscuro de la piel de estos pacientes debido a la hiperpigmentación de tipo melánica. En diversos órganos, especialmente en el corazón, se deposita hierro en forma de siderina, lo que puede conducir a insuficiencia cardíaca. La tríada clásica que caracteriza a la hemocromatosis es la presencia simultánea de cirrosis hepática, diabetes e hiperpigmentación cutánea de tipo bronceado.
La hemosiderosis generalizada secundaria se produce por un aporte aumentado de hierro por la destrucción acelerada de glóbulos rojos. En estas situaciones se acumula primero hemosiderina en el sistema fagocitario reticuloendotelial, si el aporte es mayor, se deposita en las células parenquimatosas del hígado, páncreas y miocardio.
El organismo almacena hierro en forma de hemosiderina y de ferritina. El primero es un compuesto insoluble de hidróxido férrico y una proteína denominada apoferritina. El hierro de la hemosiderina, casi en su totalidad, proviene de la hemoglobina. De los 3 a 4 gramos de hierro que normalmente se encuentra en el organismo, dos tercios se hayan asociados a hemoproteínas como la hemoglobina, la mioglobina y enzimas.
El tercio restante se halla en forma de depósito como ferritina y hemosiderina en los macrófagos del bazo, médula ósea e hígado. La cantidad de hierro que se necesita es regulado por la absorción intestinal. Así, en condiciones de déficit de hierro (deficiencias en la nutrición o hemorragias), se activan los mecanismos que aumentan la absorción intestinal. El hierro férrico proveniente de la digestión de los alimentos puede ingresar a la célula en forma «micromolecular» al ser captado en la superficie apical de los enterocitos luego de su reducción a hierro ferroso por acción de la oxidorreductasa férrica DcytB.
Una vez en estado ferroso puede entrar a la célula a través del transportador DMT 1/ Nramp 2. En el interior del enterocito el hierro puede ser almacenado como ferritina o transportado hacia fuera a través de la superficie basolateral de la célula por la ferroportina 1, reoxidado por la hefaestina (cuproproteína semejante a la ceruloplasmina pero que actúa sólo en el intestino) y finalmente, se une a la transferrina para su distribución a los tejidos.
En la membrana basolateral del enterocito, la proteína HFE modula la actividad del receptor de la transferrina en la superficie celular. La porción extracelular de la HFE debe unirse a la Beta 2 microglobina (β2M) para modular la unión del receptor TfR-1 con la transferrina 6. El interior de las vesículas de endocitosis es de carácter acídico lo que hace que el ion ferroso se libere de la apotransferrina y sea transportado al citoplasma (Fig 1). La HFE unida al receptor de la transferrina actúa como un mecanismo sensor que regula la cantidad de Fe intracelular según la cantidad de Fe del organismo 7.
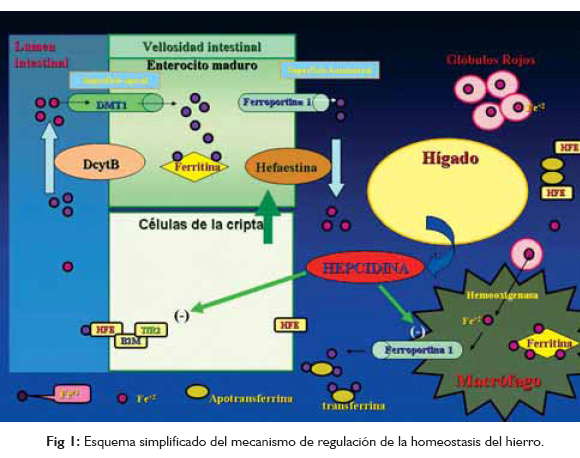
También, el hierro puede ser absorbido en forma macromolecular por fagocitosis de eritrocitos. Los macrófagos fagocitan los eritrocitos y los lisan en los fagosomas. Luego, por acción de la hemooxigenasa es liberado el hierro hemínico, el que sale del macrófago a través de un mecanismo en que participa la ferroportina y la ceruloplasmina ferroxidasa 8.
La transferrina, una β -2-globulina, transporta el hierro liberado en el catabolismo de la hemoglobina, así como también el absorbido en el intestino, para llevarlo a los sitios de almacenamiento como el hígado y el sistema retículo-endotelial, incorporándolo a la ferritina y a la hemosiderina. También, por este medio, es transportado a las células que sintetizan componentes que requieren hierro como la hemoglobina, la mioglobina y los citocromos. En el año 1996, Feder y colaboradores 9 identificaron un gen en el cromosoma 6, en la región HLA, asociado a la hemocromatosis, hoy conocido como HFE, que codifica una proteína de 343 aminoácidos (aa) y precisaron que dos mutaciones en él -C282Y y H63D dan cuenta de más del 90% de los casos de hemocromatosis hereditaria en población de origen caucásico. Este hecho significó un enorme aporte, a partir del cual se produjo una explosión de estudios que han ayudado a entender el complejo mecanismo de regulación de la absorción de Fe.
Se han identificado numerosas proteínas, algunas con actividad enzimática, otras con función de transportadoras intra o extracelulares de Fe, así como también los genes que las codifican y numerosas mutaciones en éstos, que han ido explicando cuadros de sobrecarga de hierro o anemias.
En la absorción de hierro influyen como reguladores específicos: sus depósitos, los requerimientos para la eritropoyesis y la magnitud de su ingesta. Se requerirán, por lo tanto, señales expresadas como compuestos solubles que viajen por el torrente circulatorio y comuniquen compartimentos distantes en el espacio.
Para que no exista déficit o sobrecarga de hierro, debe existir una eficiente comunicación entre las reservas de éste en el hígado, en los enterocitos duodenales y en los macrófagos, en los que la hepcidina juega un rol fundamental. La hepcidina es considerada un regulador negativo de la absorción del hierro en el intestino delgado y de su liberación por los macrófagos. Es un péptido con capacidad antimicrobiana rico en cisteínas, sintetizado en el hígado, secretado al torrente circulatorio y excretado por la orina.
La hepcidina (acrónimo de hepatic bactericidal protein sugerido por Park y colaboradores) 10 es una proteína de origen hepático
(LEAP-1: liver- expressed antimicrobial peptide 1) 11. El gen HAMP que codifica la hepcidina se encuentra localizado en la zona cromosómica 19q13. Se la ha definido como una hormona que podría ser equivalente con respecto al hierro, de lo que la insulina es al azúcar, pero aún se desconocen, o no se han descrito, sus receptores 12.
Primero se la aisló en orina y caracterizó como una proteína de origen hepático con propiedades antimicrobianas y antifúngicas 13, poco después se la correlacionó con el metabolismo del Fe 14. Hoy se sabe que esta proteína se encuentra sobreexpresada en las anemias inflamatorias y deficitarias que se acompañan con sobrecarga de hierro 15. Se la considera responsable del control de la absorción intestinal de Fe y su utilización posterior por los macrófagos. El gen que la codifica se encuentra en el cromosoma 19, consta de 3 exones y 2 intrones. Los exones codifican un primer prepropéptido de 84 aminoácidos (aa). Por acción enzimática se obtienen las formas reactivas C-terminal de 20, 22 y 25 aa. Esta última es la forma más activa de hepcidina, con una estructura que incluye 4 puentes disulfuros y un alto contenido de cisteína.
La hepcidina es sintetizada por los hepatocitos de los sinusoides estimulados ya sea por una elevación de la saturación de transferrina, o por la acción de citoquinas activadas por microorganismos que participan en la respuesta inmune e inflamatoria 16.
Un modelo de acción de la hepcidina propone que un aumento en el ingreso hepático de Fe unido a transferrina tras su unión a los receptores TfR2, aumenta su síntesis y secreción hepática. Una vez en el plasma la hepcidina interactúa con el complejo B2MHFE- TfR2 y frena la absorción de Fe por las células de la cripta duodenal y aumenta la retención de Fe por los macrófagos. Las células de la cripta duodenal comienzan un proceso de maduración a enterocitos que produce una menor expresión de transportadores celulares de Fe, resultando por consiguiente, una disminución de la absorción dietaria de Fe 17.
En este modelo, la deficiencia relativa de hierro de los enterocitos maduros y el aumento de la absorción intestinal de hierro se atribuyen a la interacción anormal entre los receptores de la transferrina TfR1 y una proteína HFE mutada en las células de las criptas.
Fraser y colaboradores 18 señalan que el hígado es capaz de detectar cambios de los niveles de transferrina diférrica (Tf) mediante los receptores HFE/TfR1 y TfR2, lo que modula la expresión del gen Hepcidina. Así, un cociente Tf/TfR disminuido, reduce la producción de hepcidina y, por el contrario, una razón mayor aumenta la producción de esta hormona hepática. En condiciones normales esta interacción aumentaría los depósitos de hierro dentro del enterocito mediante la captación de hierro de la transferrina plasmática, modularía la expresión de DMT1 y de ferroportina1 en la membrana basal y aumentaría así la concentración de las mismas en los enterocitos maduros, y por lo tanto, la absorción de hierro y su liberación hacia el plasma. Las personas con genotipo C282Y/C282Y tienen una proteína HFE que es incapaz de interaccionar con el TfR1, lo que lleva a una deficiencia de hierro en los enterocitos de las criptas, y por lo tanto, a un aumento en la expresión y funcionamiento de DMT1 y ferroportina1 que producirá una exagerada absorción de hierro y su liberación al plasma, independientemente de los requerimientos de hierro para mantener la eritropoyesis.
Cuando la sideremia es elevada, la síntesis de hepcidina aumenta disminuyendo la liberación de hierro de los enterocitos y de los macrófagos. Cuando la sideremia desciende, también descienden los niveles de hepcidina y de este modo aumenta la liberación de hierro. Aunque no se conoce el estímulo directo que controla la expresión de la hepcidina, muy probablemente las proteínas HFE, hemojuvelina y TfR2 jueguen un papel importante en él. Aun en presencia de un gen HAMP normal, una proteína HFE mutada alteraría la síntesis de la hepcidina en los hepatocitos y produciría la liberación descontrolada de hierro desde los enterocitos duodenales y los macrófagos.
La expresión de la hepcidina se correlaciona significativamente con los niveles de ferritina sérica en personas sanas 19. La excreción de hepcidina se correlaciona bien con los niveles de ferritina sérica. La excreción urinaria de hepcidina se encuentra aumentada en personas con sobrecarga de hierro y enfermedades infecciosas o inflamatorias. Se ha informado que in Vitro, el mRNAhepcidina es inducido fuertemente por la interleuquina – 6 (IL-6), pero no por IL-1 o factor de necrosis tumoral alfa (TNF-alpha). La relación que se ha establecido entre la inducción de hepcidina e inflamación, apoya la hipótesis que le atribuye un papel central en la anemia asociada a procesos inflamatorios 20.
La hepcidina en el torrente circulatorio tiene como función inhibir, tanto la absorción duodenal del Fe, como la liberación de este metal desde los macrófagos. Las citoquinas que median la respuesta inmune modifican la liberación de hepcidina desde el hígado, y por lo tanto, la disponibilidad de hierro, atribuyéndosele a este mecanismo la etiología de la anemia inflamatoria ligada a enfermedades crónicas por carencias de Fe 21, 22. La anemia que aparece como consecuencia de una enfermedad crónica se produce por la hiperactividad de la hepcidina. La proteína se activa en el proceso inflamatorio y forma parte de la lucha frente a la infección, mediante la inactivación directa de patógenos y reducción de la carga de hierro circulante. La retención del hierro priva a los patógenos de un nutriente necesario para su proliferación, pero paralelamente altera el desarrollo de hematíes, provocando anemia 22.
Los adenomas hepáticos producen altos niveles mRNA – hepcidina, demostrando con ello que ella juega un papel importante en la patogenia de las anemias asociadas a las enfermedades crónicas 23. Se ha descripto la existencia de adenomas hepáticos que segregan hepcidina, que producen anemias hipocrómicas de tipo inflamatorio, refractarias al tratamiento con Fe. La extirpación del tumor normaliza el cuadro 24. La hepcidina participa en la patogénesis de la hemocromatosis tipo 1 asociada a mutaciones del gen HFE, lo que origina una disminución de la cantidad de proteína HFE, que a su vez determina una menor producción de hepcidina, por lo que permite suponer que ambos genes están de alguna manera ligados 12. La hepcidina también puede modular la expresión de otros genes en otras enfermedades con sobrecarga de hierro, no relacionadas con el gen HFE 25.
Se ha visto en familias no relacionadas entre sí, portadoras de la variedad juvenil de hemocromatosis. Presentan dos mutaciones de genotipo heterocigoto en el gen hepcidina 26, lo que ha hecho designar a este tipo de hemocromatosis, subtipo 2, para diferenciarla de la variedad juvenil asociada a un defecto genético localizado en la porción cromosómica 1q21 (Tabla I). El proceso de absorción de Fe, la hiperpigmentación cutánea y su control, es un mecanismo complejo.
Existen importantes interacciones entre distintos factores y proteínas algunas de las cuales ya se han identificado, pero cuyas funciones se desconocen. La hemojuvelina modula la expresión de la hepcidina. La hemojuvelina regula el almacenamiento del Fe en el organismo, de modo que mutaciones en el gen que la codifica, originan una variedad juvenil de hemocromatosis 27,28.
Frente a estos nuevos conocimientos sobre el mecanismo de homeostasis del Fe, que cursan con hiperpigmentación de piel 1 y enfermedades que producen alteraciones del mismo, se puede plantear que la insuficiente producción de hepcidina por el hígado altera la absorción intestinal de Fe, provoca su sobrecarga y hemocromatosis, y por el contrario, una sobreproducción de hepcidina lleva a una menor disponibilidad de Fe y provoca anemia hipocrómica, todo lo cual puede llegar a tener una gran trascendencia, ya que se ha postulado que su utilización, de sus agonistas o de sustancias que estimulen su producción, pueden constituir una forma de tratamiento de enfermedades primarias o secundarias que se acompañan de sobrecarga de Fe (siderosis) y de la hiperpigmentación asociada, y por otra parte, el uso de antagonistas de la hepcidina podría utilizarse para el tratamiento de las anemias asociadas a procesos crónicos que son resistentes al tratamiento con eritropoyetina 24.
AGRADECIMIENTOS
Este trabajo fue realizado parcialmente con fondos de la Fundación para Estudios Biomédicos Avanzados (FEBA). Proyecto 503.
1. Wolff C, Armas R, Frank J y Poblete P. Mutations of hemochromatosis gene in volunteer blood donors and Chilean porphyria cutanea tarda patients. Medicina 2006; 66 (5): 421-426.
2. Fleming DJ, Jaques PF, Tucker KL, Massaro JM, D’Agostino RB, Wilson PW, y Wood RJ. Iron status of the free-living, elderly Framingham heart study cohort: an iron-replete population with a high prevalence of elevated iron stores. Am J Clin Nutr 2001; 73: 638-646.
3. Troisier M. Diabète sucre. Bull Soc Anat Paris 1871; 44; 231-235.
4. Von Recklinghausen FD. Uber Haemochromatose. Tegeblatt Versammlung Dtsche Naturforscher Arzte Heilgderberg 1889; 62: 324-325.
5. Merryweather-Clarke AT, Pointon JJ, Shearman JD y Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275 -278.
6. Pietrangelo A. Hereditary Hemochromatosis – A new look at an old disease. N Engl J Med 2004; 350: 2383-2397.
7. Bacon B. Hemochromatosis: Diagnosis and management. Gastroenterology 2001; 120: 718-725.
8. Gambling L, Danzeisen R, Gair S, Lea RG, Charania Z, Solanki N, Joory KD; Srai SK y McArdle SJ. Effect of iron deficiency on placental transfer of iron and expression of iron transport proteins in vivo and in vitro. Biochem J 2001; 356: 883-889.
9. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, y Wolff RK. A novel MHC class-1-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-409.
10. Park CH, Valore EV, Warin AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesied in the liver. J Biol Chem 2001; 276: 7806-7811.
11. Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VL, Livesey KJ, Tanphaichitr V, Rochette J y Robson KJ. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet 2003; 12: 2241-2247.
12. Nicolas G y Kahn A. Hepcidin, the conductor of iron homeostasis. Presse Med 2003; 32: 1395-1396.
13. Krause A, Neitz S, Mägert HJ, Schultz A, Forssmann WG, Schulz-Knappe P y Aderman K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Letters 2000; 480: 147-150.
14. Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P y Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276: 7811-7819.
15. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102: 783-788.
16. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A y Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101: 2461.
17. Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A y Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 2001; 98: 8780-8785.
18. Frazer DM y Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis 2003; 30: 288- 297.
19. Gehrke SG, Kulaksiz H, Herrmann T, Riedel HD, Bents K, Veltkamp C y Stremmel W. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to the serum transferrin saturation and to nontransferrin- bound iron. Blood 2003; 102: 371-376.
20. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A y Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003; 101: 2461-2463.
21. Dallalio G, Fleury T y Means RT. Serum hepcidin in clinical specimens. Br J Haematol 2003; 122: 996-1000.
22. Roy CN, Custodio AO, de Graaf J, Schneider S, Akpan I, Montross LK, Sanchez M,Gaudino A, Hentze MW, Andrews NC y Muckenthaler MU. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide- induced inflammation in mice. Nat Genet 2004; 36: 481-485.
23. Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI y Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 2002; 100: 3776-3778.
24. Del Castillo Rueda A, López-Herce Cid JA y De Portugal Álvarez J. Hemocromatosis Hemocromatosis hereditaria. Diagnóstico clínico: manifestaciones precoces, procesos relacionados y formas atípicas. An Med Interna (Madrid) 2002; 19: 251-256.
25. Bomford A. Genetics of haemochromatosis. Lancet 2002; 360: 1673-1681.
26. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D y Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003; 33: 21-22.
27. Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H, Cox TM, Cazzola M, Piperno A, Gimenez-Roqueplo AP, Grammatico P, Volinia S, Gasparini P y Camaschella C. Spectrum of hemojuvelin gene mutations in 1q-linked juvenil hemochromatosis. Blood 2004; 103: 4317- 4320.
28. Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR y Goldberg YP. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 2004; 36: 77-82.
Referencias
REFERENCIAS
1. Wolff C, Armas R, Frank J y Poblete P. Mutations of hemochromatosis gene in volunteer blood donors and Chilean porphyria cutanea tarda patients. Medicina 2006; 66 (5): 421-426.
2. Fleming DJ, Jaques PF, Tucker KL, Massaro JM, D’Agostino RB, Wilson PW, y Wood RJ. Iron status of the free-living, elderly Framingham heart study cohort: an iron-replete population with a high prevalence of elevated iron stores. Am J Clin Nutr 2001; 73: 638-646.
3. Troisier M. Diabète sucre. Bull Soc Anat Paris 1871; 44; 231-235.
4. Von Recklinghausen FD. Uber Haemochromatose. Tegeblatt Versammlung Dtsche Naturforscher Arzte Heilgderberg 1889; 62: 324-325.
5. Merryweather-Clarke AT, Pointon JJ, Shearman JD y Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275 -278.
6. Pietrangelo A. Hereditary Hemochromatosis – A new look at an old disease. N Engl J Med 2004; 350: 2383-2397.
7. Bacon B. Hemochromatosis: Diagnosis and management. Gastroenterology 2001; 120: 718-725.
8. Gambling L, Danzeisen R, Gair S, Lea RG, Charania Z, Solanki N, Joory KD; Srai SK y McArdle SJ. Effect of iron deficiency on placental transfer of iron and expression of iron transport proteins in vivo and in vitro. Biochem J 2001; 356: 883-889.
9. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, y Wolff RK. A novel MHC class-1-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-409.
10. Park CH, Valore EV, Warin AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesied in the liver. J Biol Chem 2001; 276: 7806-7811.
11. Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VL, Livesey KJ, Tanphaichitr V, Rochette J y Robson KJ. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet 2003; 12: 2241-2247.
12. Nicolas G y Kahn A. Hepcidin, the conductor of iron homeostasis. Presse Med 2003; 32: 1395-1396.
13. Krause A, Neitz S, Mägert HJ, Schultz A, Forssmann WG, Schulz-Knappe P y Aderman K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Letters 2000; 480: 147-150.
14. Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P y Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276: 7811-7819.
15. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102: 783-788.
16. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A y Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101: 2461.
17. Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A y Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 2001; 98: 8780-8785.
18. Frazer DM y Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis 2003; 30: 288- 297.
19. Gehrke SG, Kulaksiz H, Herrmann T, Riedel HD, Bents K, Veltkamp C y Stremmel W. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to the serum transferrin saturation and to nontransferrin- bound iron. Blood 2003; 102: 371-376.
20. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A y Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003; 101: 2461-2463.
21. Dallalio G, Fleury T y Means RT. Serum hepcidin in clinical specimens. Br J Haematol 2003; 122: 996-1000.
22. Roy CN, Custodio AO, de Graaf J, Schneider S, Akpan I, Montross LK, Sanchez M,Gaudino A, Hentze MW, Andrews NC y Muckenthaler MU. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide- induced inflammation in mice. Nat Genet 2004; 36: 481-485.
23. Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI y Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 2002; 100: 3776-3778.
24. Del Castillo Rueda A, López-Herce Cid JA y De Portugal Álvarez J. Hemocromatosis Hemocromatosis hereditaria. Diagnóstico clínico: manifestaciones precoces, procesos relacionados y formas atípicas. An Med Interna (Madrid) 2002; 19: 251-256.
25. Bomford A. Genetics of haemochromatosis. Lancet 2002; 360: 1673-1681.
26. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D y Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003; 33: 21-22.
27. Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H, Cox TM, Cazzola M, Piperno A, Gimenez-Roqueplo AP, Grammatico P, Volinia S, Gasparini P y Camaschella C. Spectrum of hemojuvelin gene mutations in 1q-linked juvenil hemochromatosis. Blood 2004; 103: 4317- 4320.
28. Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR y Goldberg YP. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 2004; 36: 77-82.


