
Autores | Contacto
SL Rastellini *
* Cursista de Tercer Año del Curso Superior para Médicos Especialistas en Dermatología. Hospital General de Agudos “Dr. Ignacio Pirovano”.
Dr. Ricardo E. Achenbach. Profesor Regular Adjunto. Jefe de Unidad. Asignatura Dermatología.
Hospital General de Agudos “Dr. Ignacio Pirovano”. Av Monroe 3550 (1430). Ciudad Autónoma de Buenos Aires. Argentina.
E-mail: silvanaluzr@gmail.com
Recibido: 18-05-2015
Aceptado para su Publicación: 20-08-2015
Dirección
Prof. Dr. Ricardo E. Achenbach
Resumen | Palabras Claves
RESUMEN
El carcinoma de células de Merkel (CCM) es un tumor cutáneo maligno poco frecuente, de rápido crecimiento, localmente agresivo, con tendencia a dar metástasis ganglionares, a distancia y con alta tasa de recidiva local. Su etiopatogenia se desconoce, pero recientemente se ha demostrado la replicación de un virus en su interior (poliomavirus de células de Merkel), lo que ha supuesto un nuevo campo de investigación. Requiere diagnóstico precoz, tratamiento quirúrgico y radiante para mejorar la sobrevida de los pacientes. Se comunica un caso de CCM en una mujer de 72 años, a la que se le confirma el diagnóstico por las características histopatológicas e inmunohistoquímicas, debido a la forma de presentación atípica de gran tamaño, rápido crecimiento y localización infrecuente. Se efectúa una revisión de la entidad, además se describen la evolución clínica y el tratamiento instaurado, sin recidiva a siete meses de seguimiento.
PALABRAS CLAVE: Carcinoma de células de Merkel; Diagnóstico; Inmunohistoquímica; Terapéutica.
SUMMARY
The Merkel Cell Carcinoma (MCC) is a rare malignant skin tumor, fast-growing, locally aggressive, prone to lymph node and distant metastases and high rate of local recurrence. Its pathogenesis is unknown, but has recently shown a virus replication inside (Merkel cell polyomavirus) which has led to a new field of research. It requires early diagnosis and surgical treatment and radiotherapy to improve the survival of patients. CCM case is reported in a 72 years at diagnosis by the histopathologic and immunohistochemical characteristics are confirmed, due to the atypical presentation of large, fast-growing and unusual location. A review of the peculiar disease and also the clinical course and treatment instituted, without recurrence at seven months of follow-up are described.
KEY WORDS: Merkel cell carcinoma; Diagnosis; Immunohistochemistry; Therapeutics.
Artículo | Referencias
Descargar archivo PDF aquí
INTRODUCCIÓN
El carcinoma de células de Merkel (CCM), fue descrito por primera vez por Toker en 1972, bajo el nombre de carcinoma trabecular de la piel. Es un tumor cutáneo maligno poco frecuente, que representa menos del 1% de los tumores cutáneos malignos, localmente agresivo, con tendencia a dar metástasis ganglionares, a distancia y con alta tasa de recidiva local. La ultraestructura y la inmunohistoquímica son similares a las de las células neuroendócrinas. Su etiopatogenia se desconoce, pero se ha comunicado su asociación con otros tumores epiteliales y recientemente, se ha demostrado la replicación de un virus en su interior. Requiere diagnóstico precoz, tratamiento quirúrgico y radiante, para mejorar la sobrevida de los pacientes.
Se presenta el caso clínico de una paciente, a la que se le confirma el diagnóstico de CCM por las características histopatológicas e inmunohistoquímicas, debido a la forma de presentación atípica de gran tamaño, rápido crecimiento y localización infrecuente. Además se describen la evolución clínica y el tratamiento instaurado sin recidiva, a siete meses de seguimiento.
CASO CLÍNICO
Mujer de 72 años, con antecedentes de insuficiencia venosa crónica, medicada con venotónicos, que consultó a nuestro Servicio de Dermatología, derivada del Servicio de Flebología, por tumoración en región glútea derecha de tres meses de evolución, de 7×6 cm, eritemato-violácea, levemente descamativa, de bordes netos, asintomática, con nódulos que la coronaban y se ubicaban en forma intra tumoral. Éstos se palpaban duros (Fig 1). Además presentaba adenopatía inguinal derecha, dura, adherida a planos profundos, asintomática. En el resto del examen físico no se encontraron hallazgos patológicos.

Fig 1: A y B. Carcinoma de células de Merkel en región glútea derecha de tres meses de evolución. Se observa tumoración de 7×6 cm, eritemato-violácea, levemente descamativa, de bordes netos, asintomática, con nódulos que la coronan y se ubican en forma intratumoral, duros.
Se solicitó una ecografía de piel y partes blandas de la tumoración glútea, donde se observaron formaciones nodulares múltiples, hipoecogénicas, de contornos definidos, ubicados en el espesor del tejido celular subcutáneo, siendo la mayor de 49×22 mm, localizada a 2,5 mm de la superficie de la piel.
Los resultados del laboratorio de rutina arrojaron parámetros normales, excepto una medición del colesterol total de 278 mg%.
En primera instancia, mientras se aguardaba el resultado de la biopsia realizada por punch de la lesión glútea, se plantearon los siguientes diagnósticos diferenciales: linfoma B, metástasis de cáncer ginecológico, melanoma amelanótico y carcinoma espinocelular. A tal fin se realizó un examen ginecológico, que incluía Papanicolau, colposcopía, mamografía y ecografía ginecológica, con hallazgos acorde al sexo y edad; una ecografía abdominal y radiografía de tórax sin hallazgos patológicos; los marcadores para alfafetoproteína y CA 125 fueron negativos; el proteinograma electroforético con valores fisiológicos.
El estudio anatomopatológico reveló la presencia de CCM: se halló en la dermis una proliferación atípica de células pequeñas de núcleos ovoides, hipercromáticos, con muy escaso o nulo citoplasma, dispuestas en patrón difuso; además se observó otro sector donde la lesión ulceraba la epidermis, visualizándose numerosas embolias vasculares y áreas de necrosis del tumor (Fig 2); con marcadores inmunohistoquímicos positivos para citoqueratina 20 (patrón en “dot”), AE1 – AE3 (antígeno epitelial específico de membrana), cromogranina y sinaptofisina (Fig 3). El Ki67 fue del 80% (lo que indica actividad proliferativa elevada). Asimismo los marcadores para ACL (antígeno común leucocitario), TTF1 (factor de transcripción tiroidea-1), CK5, CK7 (citoqueratinas 5 y 7), HMB45, CD20 y CD3 (anticuerpos monoclonales) fueron negativos.
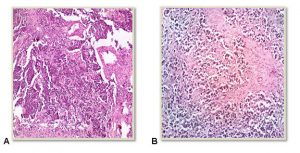
Fig 2: histopatología de CCM. HyE, 10X. Corresponde a la primera biopsia incisional. A. Se observa en dermis una proliferación atípica de células pequeñas, de núcleos ovoides, hipercromáticos, con muy escaso o nulo citoplasma, dispuestas en patrón difuso. B. La lesión ulcera la epidermis. Se observan embolias vasculares y áreas de necrosis tumoral.

Fig 3: histopatología del CCM. Tinciones inmunohistoquímicas. A. AE1 – AE 3 (antígeno epitelial específico de membrana). B. Citoqueratina 20. Obsérvese el patrón en “dot” (gota).C. Cromogranina. D. Sinaptofisina.
La tomografía por emisión de positrones (PET) corporal total informó: masa adenomegálica hipermetabólica sobre la cadena ilíaca externa y la cadena inguinal derecha; además se visualizó una imagen de partes blandas hipermetabólica sobre la región glútea derecha, en planos superficiales.
Se interconsultó con el servicio de Cirugía General del hospital, donde le realizaron el vaciamiento ganglionar ilioinguinal derecho, con preservación del músculo fascia lata, más la resección local amplia del tumor glúteo, con márgenes negativos.
Concomitantemente fue evaluada por el servicio de Oncología, efectuando tratamiento radiante en el lecho quirúrgico y de los ganglios linfáticos regionales, con dosis diaria de 2 Gy, llegando a una dosis total de 54 Gy por un lapso desde el 14/10/2014 al 20/11/2014, con buena tolerancia al tratamiento.
Un mes después, la paciente concurrió al control dermatológico, donde al examen físico se constató linfedema del miembro inferior derecho y se palpó una tumoración en región inguinal izquierda, dura, adherida a planos profundos, asintomática, por lo que se realizó TAC (tomografía axial computada) corporal total, que informó: como dato positivo en abdomen y pelvis un ganglio inguinal izquierdo, de aproximadamente 10 mm de diámetro. Además se observó imagen de hipodensidad, con densidad líquida y paredes definidas, localizada en la profundidad de la fascia del músculo oblicuo derecho, con extensión a región inguinal, siendo su diámetro máximo de aproximadamente 11x4x6 cm, que podrían vincularse, en primera instancia, a colección. Por tal motivo, se realizó extirpación quirúrgica de dicha adenopatía inguinal izquierda y al examen histopatológico se diagnosticó adenitis reactiva inespecífica, por lo que fue negativo para neoplasia. Actualmente, a los siete meses de seguimiento, la paciente sigue concurriendo a nuestro servicio para realizar los controles indicados mensualmente, sin evidencia de recidiva local ni a distancia.
HISTORIA
Friedrich Sigmund Merkel fue uno de los más brillantes anatomistas del siglo XIX y principios del XX, que en el transcurso de su vida hizo importantes contribuciones al conocimiento de la anatomía e histología. En 1875 Merkel describió unas células peculiares, localizadas en la capa basal de la epidermis, en las que penetraban fibras nerviosas, cuya vaina de mielina terminaba en una protuberancia por fuera de la célula, no eran dendríticas y no participaban en la “queratinopoyesis” y por lo tanto, no descamaban. Merkel designó estas células, nunca vistas hasta entonces, con el nombre de “Tastzellen” (células del tacto) y más tarde se las denominó células de Merkel. 1,2
El origen de estas células aún está en discusión, aunque la teoría más aceptada en la actualidad sugiere que surgen durante la embriogénesis, derivada de la cresta neural o bien de células epidérmicas pluripotentes. Su distribución topográfica, de forma irregular, muestra un predominio en la piel glabra de los pulpejos, área centrofacial y también ha sido descrita en el lecho ungular, en las mucosas nasal y bucal, encía, borde bermellón de los labios y paladar, así como también, en la vaina epitelial externa del folículo piloso. Son abundantes en los discos pilares, a los que en la actualidad se los reconoce como estructuras mecanorreceptoras de adaptación lenta. Se demostró que la característica más sobresaliente de la célula de Merkel, es la presencia en su citoplasma de vesículas granuladas similares a las observadas en las células neuroendócrinas, que poseen en su interior un gránulo electrodenso, separado de la membrana vesicular por un espacio electrolúcido y que miden entre 100 a 400 mm de diámetro, lo que determinó que la célula de Merkel se integre al llamado sistema APUD (“Amine Precursor Uptake and Decarboxylation”), ya que, comparten múltiples propiedades con otros elementos del sistema, como su origen en la cresta neural, su argentafinidad, la presencia de esterasas y gránulos intracitoplasmáticos limitados por membrana, así como, la producción de hormonas y el desarrollo de síndromes paraneoplásicos. 1,3
Respecto de la función de la célula de Merkel, en su descripción original el autor la consideraba como un transductor específico del estímulo mecánico a la información neural. En realidad, hasta la fecha esta hipótesis no ha podido ser confirmada fehacientemente puesto que, si bien se la ha visto formando parte de terminaciones corpusculares de reconocida función mecanorreceptora, no se ha aseverado su función transductora.
La proliferación neoplásica maligna de las células de Merkel corresponde al carcinoma de células de Merkel (CCM), que fue descrito por primera vez por Toker en 1972, bajo el nombre de carcinoma trabecular de la piel, por su carácter infiltrante y el patrón histopatológico.
En 1978, Tang y Toker observaron gránulos en las células tumorales semejantes a los de la célula de Merkel, un hallazgo que sugiere que el tumor deriva de éstas. Desde entonces ha recibido diversas denominaciones, tales como: “neoplasia cutánea de células de Merkel”, “APUDOMA cutáneo”, “carcinoma neuroendócrino de la piel”, “carcinoma primitivo de la piel de células pequeñas”, “carcinoma indiferenciado de la piel”, “tumor cutáneo maligno de la célula de Merkel”, “carcinoma cutáneo primitivo de células pequeñas con diferenciación endócrina”, “merkeloma”, “tumor de Merkel” y “neuroblastoma del adulto”. 2,3,4
En nuestro país, el primer caso de CCM fue descripto por Magnin y col en 1982. 2,5
EPIDEMIOLOGÍA Y FACTORES ETIOPATOGÉNICOS
EPIDEMIOLOGÍA
El CCM es un tumor poco frecuente, menos del 1% de todos los tumores cutáneos malignos, 100 veces menos frecuente que el melanoma. La incidencia ha ido en aumento en los últimos años a más de 1.000 casos por año en los Estados Unidos, por lo que se lo ubicó recientemente como el segundo cáncer cutáneo primario, luego del melanoma, causante de muerte. 3,6
Su epidemiología varía de acuerdo a las series que se revisen, la mayoría señalan predominio en varones, variando la proporción de 1,4-1,5:1 a 2-3:1 y otros autores, sin embargo, informan predominio en mujeres; con mayor incidencia entre la sexta y la octava décadas de la vida (media: 69 años) y sólo el 5% antes de los 50 años. 3,4,6,7 En nuestro caso la paciente tiene 72 años. Sin embargo, la enfermedad ocurre a una edad inferior en sujetos inmunodeprimidos. Es más frecuente en caucásicos y pacientes con antecedentes de carcinoma basocelular y carcinoma espinocelular, con los que comparte muchos de los factores de riesgo. 6,7
FACTORES ETIOPATOGÉNICOS
Aunque la etiología es dudosa, hay varios factores que contribuyen a su patogenia, entre ellos: la exposición crónica a la radiación ultravioleta y la inmunosupresión, que se proponen como principales factores etiopatogénicos implicados en el desarrollo de este carcinoma.
En cuanto a la exposición crónica a la radiación ultravioleta, hay una serie de evidencias que la apoyan:
- La mayor incidencia en pacientes de raza blanca y en localizaciones foto-expuestas. Varios autores han encontrado relación entre el índice de radiación UVB y la incidencia de CCM.
- La marcada asociación de los CCM con otros procesos cutáneos relacionados con el daño actínico, tales como: queratosis actínica, carcinoma epidermoide, carcinoma basocelular o enfermedad de Bowen.
- La aparición de CCM en pacientes en tratamiento con psoralenos y radiación ultravioleta A (PUVA).
- La publicación de una mutación en el gen p53 típicamente relacionada con la radiación ultravioleta B, similar a la descrita en el carcinoma basocelular y en el carcinoma epidermoide. 3,4,7,8,9
No obstante, dado que el CCM en ocasiones se presenta en áreas no foto-expuestas como en nuestro caso, se postulan otros factores etiológicos entre los que se destaca la inmunosupresión. Entre los hallazgos que avalan esta hipótesis se encuentran:
- La demostración de casos de remisión espontánea, tanto del tumor como de las metástasis linfáticas regionales, presumiblemente inmunomediada. 9,10
- La asociación del CCM con situaciones de inmunodeficiencia, como son:
- Secundaria a la administración de tratamiento inmunosupresor, en relación con otros procesos concomitantes: enfermedades autoinmunes, fracaso renal crónico, sarcoidosis pulmonar, trasplante renal y cardíaco.
- En relación con otras enfermedades concomitantes de por sí inmunosupresoras: pacientes infectados con el VIH, neoplasias hematológicas como: leucemia, mieloma múltiple, linfoma no Hodgkin.
- Además, el CCM puede estar asociado con otros tumores no cutáneos en un 25% de los pacientes, por ejemplo de mama, ovarios, cabeza y cuello, que pueden favorecer una inmunosupresión secundaria, bien por los fármacos inmunosupresores o por las neoplasias en sí mismas. 7,9,10
La exposición al arsénico y su coincidencia con polidisplasia con hipoplasia dérmica focal y la enfermedad de Cowden, también han sido señaladas en diversas publicaciones. 10
En el año 2008, Feng y col describieron la presencia de un nuevo poliomavirus en 8 de 10 pacientes con CCM, al que lo llamaron poliomavirus de células de Merkel (MCV). Dado que el contacto con el virus precedería a la aparición del tumor, se cree que contribuiría a su patogenia. 2,4,11
Estudios genéticos recientes han demostrado deleción del brazo corto del cromosoma 1 (1p36) y la pérdida de la capacidad heterocigota en el cromosoma 3p21, 2 lo que también se observa en el carcinoma de pulmón, que se encuentra incluido como diagnóstico diferencial, pero se trata de anomalías genéticas de baja resolución y hasta la actualidad, no se ha identificado un gen supresor tumoral o un oncogén implicados de manera concluyente en el CCM. 9
PRESENTACIÓN CLÍNICA
El CCM se localiza con preferencia en áreas descubiertas (foto-expuestas): en el 65% de los casos afecta la cabeza y el cuello; en el 18%, extremidades superiores; en el 13%, extremidades inferiores y con menor frecuencia en el tronco. 2,3,4 También se ha informado compromiso de otros sitios de piel protegida del sol, incluyendo vulva, pene, faringe, mucosas nasal y bucal y glúteos 8,9,10, como es en el caso de nuestra paciente.
En la mayoría de los casos se presenta como un tumor cutáneo primario, único, en general asintomático, su color varía desde el mismo tono de la piel al castaño rojizo o violáceo; suele ser sésil, cupuliforme o en placa, de consistencia firme y superficie lisa, puede acompañarse de una ulceración superficial con componente hemorrágico y su tamaño varía de unos pocos milímetros a varios centímetros (generalmente, menor de 2 cm). 2,3,4,7,8 Se han descrito algunas formas gigantes, de hasta 23 cm. 9 En el caso de nuestra paciente el diámetro mayor era de 7 cm.
Generalmente su crecimiento es rápido de uno a tres meses. La mayor parte de los pacientes (76-89%) debutan como enfermedad localizada al diagnóstico; con afectación ganglionar entre el 10-18% y con metástasis a distancia entre un 1-2%. Sin embargo, se trata de un tumor agresivo, con una tasa de recurrencia de entre el 33 y el 44%, con afectación ganglionar lo corregional hasta en el 55-60% de los casos y metástasis a distancia hasta en el 33% a piel, hígado, pulmón, hueso y cerebro. 7,9,10,12
Aunque es 40 veces menos común que el melanoma, tiene una mortalidad mayor (33%) que éste (15%) a los tres años. 9
En ocasiones se confunden con carcinomas basocelulares, carcinomas espinoceluñares, melanomas amelanóticos, hemangiomas y especialmente linfomas cutáneos. 4,5,7,9,10,12 Con frecuencia, el CCM no se incluye en el diagnóstico clínico inicial, como ocurrió en nuestro caso.
Heath y col, en un estudio de cohorte prospectivo de 195 pacientes, describieron que la mayoría (88%) eran asintomáticos, de rápido crecimiento, coloración roja a rosada en un 56%. La localización se relacionaba con la exposición solar previa en un 81% de los tumores primarios y la mayor cantidad de pacientes (90%) tenía más de 50 años. Este grupo creó un acrónimo que caracteriza clínicamente a estos tumores: AEIOU (Asintomático; Evolución con rápido crecimiento; Inmunosupresión; Older: más de 50 años; UVB expuestos). 4
Nuestra paciente presentaba tres de las características clínicas del acrónimo: tumor asintomático, evolución con rápido crecimiento y 72 años de edad.
Se describen tres formas de presentación clínica:
- Tumor solitario (como en el caso de nuestra paciente).
- Múltiples tumores en un área única (semejante a la satelitosis del melanoma).
- Múltiples tumores de distribución amplia, también denominada “merkeliomatosis cutánea”. 3,10,12
A su vez se han descrito diversas formas clínicas de presentación atípica, como por ejemplo: en forma de una mínima ulceración en la punta de la nariz, nódulos subcutáneos en la región inguinal, tejido de granulación en el pie de un adolescente, una lesión en el pezón de una mujer o una placa eritematosa que semeja un angiosarcoma. 10,12
HISTOPATOLOGÍA
El estudio histopatológico se caracteriza por una proliferación de pequeñas células monomorfas, con núcleos grandes, redondos, irregulares y vesiculares, con la cromatina granular finamente dispuesta en su interior y múltiples nucléolos excéntricos y pequeños, con alto grado de mitosis. El citoplasma es escaso y basófilo. Con frecuencia presenta apoptosis.
El CCM es una proliferación celular que se localiza en la dermis; si bien no invade la epidermis, en algunos casos ésta puede aparecer ulcerada por compresión. También se ha descrito que puede comprometer la hipodermis y los vasos sanguíneos. El estroma puede contener vasos proliferantes y células endoteliales prominentes.
Al microscopio electrónico las células del CCM son similares a las células de Merkel normales. El estudio ultraestructural es el primer medio por el que este tumor se puede diferenciar de otros tumores, poco diferenciados de células pequeñas. En el citoplasma se encuentran los gránulos electrodensos de halo claro característicos, neurosecretorios, como también agregados paranucleares de filamentos intermedios compuestos sólo por citoqueratinas. 9,10,12
Los patrones de crecimiento histopatológico se dividen en tres:
- Sólido o nodular, que se caracteriza por “colgajos”, “sábanas” y grandes grupos intradérmicos de células pequeñas e hipercromáticas, sin diferenciación glandular. A menudo muestran áreas de necrosis y se asemejan a otros tumores indiferenciados, de células pequeñas.
- Difuso, es el más frecuente (50% de los casos), en el que las células se distribuyen de modo uniforme “en manto”, caracterizado por grandes nidos sólidos de células tumorales separados por finos tractos de tejido conectivo y focos de necrosis. Suelen presentar un número elevado de mitosis. Hay un infiltrado linfocitario dentro y alrededor del tumor. Los tumores que adoptan este patrón de crecimiento, pueden situarse próximos a los anexos y conectarse con la epidermis. Este patrón presentaba nuestra paciente.
- Trabecular, descrito por primera vez por Toker en 1972, es el patrón mejor diferenciado y el menos frecuente de los tres, observándose en menos de la cuarta parte de los casos publicados. Las células están dispuestas en un patrón “organoide” de trabéculas sólidas, irregulares y anastomosadas que pueden exhibir formaciones pseudoglandulares. En esta variante, el índice mitótico es escaso o moderado. 2,3,4,9,12
INMUNOHISTOQUÍMICA
El uso de tinciones inmunohistoquímicas basadas en el empleo de anticuerpos monoclonales, ha aumentado en gran medida la facilidad y la especificidad del diagnóstico del CCM. Éste expresa marcadores de origen epitelial y de tipo neuroendócrino. A saber:
- Citoqueratina-20 (CK 20): es la más útil de estas tinciones, positiva en el 97% de los casos.8 Es la proteína de los filamentos intermedios del CCM. Además se expresa en los adenocarcinomas de colon, estómago y páncreas. En la piel, sin embargo, su expresión se limita a las células de Merkel. Un patrón de “puntillado perinuclear” (“dots” paranucleares) de citoqueratina es esencialmente patognomónico del CCM. La CK 20 también se usa en la detección de micrometástasis ganglionares, fundamentalmente en el ganglio centinela. Ésta fue positiva para la muestra tomada en el caso que presentamos.
- Antígeno epitelial de membrana (AE1/AE2y): son proteínas presentes en la mayoría de las células epiteliales. Sin embargo, no es específica debido a que también se expresa en células plasmáticas y en algunos linfomas. Presente en la mayoría de los tumores de origen epitelial, meningiomas y mesoteliomas. En nuestro caso estuvo presente.
- Anticuerpos anti-CAM5.2 (anticuerpos anticitoqueratina de bajo peso molecular y neurofilamentos): detectan múltiples epítopes de citoqueratinas humanas, reaccionan tanto con el CCM como con el carcinoma pulmonar a células pequeñas. Aunque es útil, como herramienta de rastreo inicial para detectar tumores de origen escamoso, su falta de selectividad en comparación con la CK-20 implica que no puede utilizarse, para el diagnóstico definitivo del CCM.
- Marcadores neuroendócrinos: cromogranina, sinaptofisina, enolasa neuroespecífica: estos son positivos en el CCM y en otros neoplasmas derivados de tejidos nerviosos o neuroendócrinos. En algunos casos se liberan de los carcinomas neuroendócrinos, entre ellos el CCM y pueden detectarse en sangre. La cromogranina y la sinaptofisina arrojaron un resultado positivo en nuestro caso. No se midió enolasa neuroespecífica. 9,13,14
Estas técnicas facilitan el diagnóstico diferencial, principalmente entre el CCM y el carcinoma de pulmón de células pequeñas metastásico a piel.
En relación con el empleo de la CK20, ha adquirido una gran importancia el TTF-1 (factor de transcripción tiroidea-1), factor de transcripción nuclear expresado por células tiroideas y de pulmón y por la mayoría de los carcinomas epidermoides. Este marcador se expresa también en adenocarcinomas (72.5%), carcinomas de células pequeñas (83% -100%), tumores carcinoides atípicos (100%) y carcinomas neuroendocrinos (75%). No es expresado, sin embargo, por los CCM.
Los datos iniciales publicados por Byrd-Gloster y col, muestran que el TTF-1 es expresado por el 97% de los carcinomas de células pequeñas de pulmón, pero no por los CCM. En el mismo estudio, la CK 20 etiqueta el 76% de los CCM, mientras que es expresada únicamente por el 3% de los carcinomas de células pequeñas de pulmón. Otro estudio ha señalado la expresión de TTF-1 en el 82.7% de los carcinomas de células pequeñas de pulmón, en el 42% de los carcinomas de células pequeñas extra pulmonares y en el 0% de los CCM. 9,13
De este modo, una combinación de TTF-1 y CK 20 debería proporcionar la mayor sensibilidad y especificidad, para distinguir entre CCM y otros carcinomas de células pequeñas, aunque no diferencie, en estos últimos, entre un origen pulmonar o extra-pulmonar. En el caso de nuestra paciente TTF-1 fue negativo.
La CK 7 (citoqueratina 7) tiene el mismo patrón de tinción que el TTF-1, es decir, también resulta típicamente negativo en CCM y positivo en el carcinoma pulmonar a células pequeñas por lo que, en combinación con CK 20 y TTF-1, puede ayudar a diferenciar el CCM de metástasis cutáneas de un carcinoma bronquial de células pequeñas. También se expresa en las células epiteliales del pulmón, ovarios y mamas. La CK5 (citoqueratina 5) es una queratina de alto peso molecular, que se expresa en tumores escamosos. CK 7 y CK 5 resultaron negativas en nuestro caso.
En el CCM no se detectan proteína S-100, HMB-45 (anticuerpo monoclonal) y NK 1/C3, útiles para el diagnóstico del melanoma. Tampoco se aprecia expresión para integrina, proteína ácida fibrilar glial, antígeno común leucocitario (ACL), actina, vimentina, laminina ni metencefalina. Esta última es un marcador de las células de Merkel normales. ACL es una glicoproteína transmembrana con actividad tirosinfosfatasa, útil para diferenciar entre linfomas y carcinomas, ya que, es positivo en los primeros y negativo en los segundos, entre ellos el CCM. En el caso de nuestra paciente se midió HMB-45 y ACL cuyos resultados fueron negativos.
El CD20 es un marcador específico de células B y resultó ser negativo para nuestra paciente. Los anticuerpos monoclonales anti CD2, CD3, CD4, CD5, CD7 y CD8 son los marcadores más frecuentemente utilizados en el estudio de infiltrados de linfocitos T. En nuestro caso se buscó marcación con CD3, con resultado negativo.
La evaluación del índice de proliferación en el CCM medido por Ki-67, se demostró que posiblemente se correlacione con progresión de la enfermedad. La expresión de Ki-67 medida como el porcentaje de células tumorales, se asoció significativamente con un peor pronóstico en un estudio realizado por Koljonen y col y Llombart y col. 13 Muchos expertos están de acuerdo en el impacto pronóstico del elevado recuento mitótico en CCM, sin embargo, en una serie, el porcentaje de Ki-67 no parece correlacionar con el resultado (“outcome”) clínico. 14Una posible explicación de los diferentes informes de Ki-67 como un indicador pronóstico, es que los anti-Ki-67 no identifican específicamente las células en la fase mitótica del ciclo celular. 9,13,14,15
En el caso clínico que presentamos el Ki67 fue del 80%.
DIAGNÓSTICO
A pesar de lo descrito anteriormente, debido a la baja prevalencia del CCM, no se han realizado grandes estudios sobre el sistema de estadificación óptimo. Existe escasa literatura sobre el algoritmo de imágenes en pacientes con CCM. La lesión primaria es relativamente fácil de evaluar, mediante el examen clínico y la histopatología, sin embargo, la determinación de la afectación ganglionar regional es a menudo más difícil. Las opciones para la estadificación ganglionar incluyen el examen clínico, la biopsia del ganglio centinela, la disección de los ganglios linfáticos y la imagen anatómica o funcional.
Se describen las herramientas de diagnóstico en imágenes del CCM:
1. Ecografía: es una técnica muy precisa y barata en la estadificación tumoral. En lo que se refiere al carcinoma de células de Merkel, se debiera iniciar con una ecografía.
Las lesiones primarias de la piel pueden aparecer como nódulos únicos o multicéntricos, sólidos, hipoecoicos que surgen de la dermis y se extienden a la grasa subcutánea, con grados variables de transmisión acústica posterior. Las características ecográficas del CCM parecen ser similares a los tumores más comunes de la piel, tales como: melanoma o carcinoma de células basales. 16,17
La ecografía tiene un papel clave en la formación de imágenes en tiempo real durante la biopsia, con aguja fina de lesiones no palpables de carcinoma de células de Merkel. A excepción de algunos casos comunicados, la biopsia por aspiración con aguja fina guiada por ultrasonido, ha sido raramente descrita en pacientes con CCM. 16
Es un desafío el realizar el diagnóstico definitivo de enfermedad metastásica, sólo con citología por aspiración con aguja fina. La citomorfología se asemeja a otros numerosos tumores como los linfomas y melanomas. Sin embargo, la aspiración con aguja fina de CCM puede proporcionar un diagnóstico preciso y fiable de lesiones metastásicas primaria o recurrente. En los pacientes con ganglios positivos demostrados, debe hacerse una imagen de cuerpo completo para detectar metástasis distantes. 16,17,18
2. Biopsia del ganglio centinela (BGC): la BGC ofrece la capacidad para detectar metástasis y micrometástasis y posteriormente, drenaje del ganglio linfático metastásico en pacientes con melanoma, carcinoma de células escamosas y CCM mediante el uso de linfogammagrafía. La BGC en pacientes con CCM parece ser una técnica de estadificación fiable, mientras que la importancia pronóstica de tumor positivo en el ganglio centinela, sigue siendo poco clara.
Según lo mencionado en varias series de casos la biopsia del ganglio centinela para el CCM, se informó afectación ganglionar en aproximadamente un tercio de pacientes, con ganglios clínicamente negativos. También se ha documentado que los pacientes con ganglios negativos histopatológicamente, han mejorado la supervivencia y redujeron las tasas de recurrencia, en comparación con los pacientes con ganglios clínicamente negativos. Sin embargo, otras series de casos comunicaron altos índices de recaída en pacientes con la biopsia del ganglio centinela negativa, manifestando así algunas dudas sobre la exactitud de esta técnica. Debido al drenaje linfático variable en enfermedades malignas cutáneas, se requiere radiotrazador o tinte para identificar el ganglio linfático centinela, pero esta técnica no identifica el 100% de los ganglios linfáticos centinelas y los resultados de la linfogammagrafía, pueden variar con el tiempo para cada individuo. 9,16,18,19
Es muy probable que esta técnica sea irrelevante para aumentar la sobrevida del paciente, tal como ocurre en el melanoma.
3. Tomografía Computada (TC): debido a la utilidad de la TC para visualizar los nódulos linfáticos de la cabeza y el cuello, así como para las metástasis nodulares en la grasa subcutánea y metástasis viscerales, varios autores propusieron que la TC es un método de imágenes fiable, para la estadificación inicial de los pacientes con carcinoma de células de Merkel.
En comparación con el músculo, las lesiones cutáneas primarias aparecen como nódulos cutáneos redondeados isodensos a ligeramente hiperdensos, que se extienden por debajo de la piel. La grasa cutánea adyacente a la lesión primaria, sugiere congestión y edema por invasión linfática. Además, la TC realzada es capaz de demostrar linfadenopatía con alta atenuación y la TC suave (soft TC), es capaz de demostrar con alta atenuación los nódulos tisulares, que a menudo son clínicamente silentes, sugiriendo metástasis focales.
La linfadenopatía mayormente se produce en el cuello, especialmente en la región parótida seguida por la axila, mediastino, retroperitoneo y la ingle. Las metástasis a distancia incluyen los ganglios linfáticos locales y retroperitoneales, el hígado, hueso, cerebro y pulmón. Las metástasis de los órganos abdominales se manifiestan como lesiones hipervasculares, con un realce en anillo. Las metástasis de tejidos blandos pueden implicar la pared torácica o abdominal, con invasión musculoesquelética. Gollub y col realizaron un estudio en 12 pacientes con CCM y mostraron la capacidad de la TC para detectar metástasis viscerales y nodales. Ellos sugirieron seguimiento con TC a los 3, 6, 12 y 18 meses después del tratamiento inicial, para descubrir la enfermedad recurrente. 16,17,18
4. Resonancia Magnética (RM): existen sólo unos pocos estudios e informes de casos, que describen la utilidad de la RM en pacientes con CCM. Éstos se describieron como tumores primarios, grandes, de la región nasosinusal y de la pared abdominal como imágenes con intensidad de señal no homogéneas en T1 y T2.
El aumento de la intensidad de señal central sobre imágenes en T2 de grandes lesiones, se describió como asociada histológicamente con necrosis central y hemorragia. En la RM las lesiones satélites linfáticas se observan como una masa reticular y subcutánea. La misma apariencia de lesiones satélites pueden observarse por TC, grandes metástasis en los ganglios linfáticos aparecen como lesiones sutiles, comprimidas, con tejido graso retenido. Además, las masas intramusculares y tumores perifaciales, se definen mejor en la RM que por la TC.
La RM en los carcinomas de células de Merkel es muy precisa, para la evaluación de las metástasis de tejidos blandos, así como la participación del cerebro y la médula ósea. La invasión del sistema nervioso central es poco frecuente; sin embargo, en caso de síntomas neurológicos, el estudio diagnóstico debe realizarse con RM. 16,17,18
5. Gammagrafía con receptor de somatostatina (GRS): la razón para la realización de GRS en pacientes con CCM, para detectar enfermedad metastásica locorregional y distante, se basa en las características neuroendócrinas del CCM. En 1992, Kwekkeboom y col presentaron datos para la efectividad de GRS en 4 pacientes con MCC. En todos ellos, en los que se detectó el tumor por TC y ecografía, también se detectó por GRS. Ellos mostraron que GRS tenía una sensibilidad igual o mayor que la TC, para obtener imágenes de CCM. 18
Sin embargo, estudios más recientes 16 observaron una limitada sensibilidad de GRS, así como, una alta tasa de falsos positivos y negativos. Se realizó una comparación entre GRS, TC y RM que mostró que el tejido evaluado por GRS está menos afectado por inflamación, edema y tejido de granulación en los sitios previamente tratados quirúrgicamente o irradiados. No obstante, existe un valor limitado significativamente, en los órganos que presenten una absorción fisiológica del radiomarcador octreotide como el hígado, las glándulas suprarrenales, el páncreas, la glándula tiroides y el bazo. Esto dificulta la detección de metástasis cerca de órganos, con una alta captación fisiológica del trazador. Además, otras enfermedades sistémicas como sarcoidosis, tuberculosis, granulomatosis de Wegener, linfoma de Hodgkin o no Hodgkin, también llevaron a resultados falsos positivos de GRS.
Por lo tanto existe un uso limitado de GRS en la evaluación diagnóstica de CCM, por ello, muchos autores no lo recomiendan como imagen de rutina. 16
6. Tomografía por Emisión de Positrones (PET) y Tomografía Computarizada por Emisión de Positrones (PET-TC): en los últimos años de medicina nuclear, especialmente la PET y PET-TC, han ganado importancia en el diagnóstico por imagen del CCM que, al ser un tumor de crecimiento rápido, se espera que las células tumorales tengan un aumento de la glicólisis. El 18F-fluodesoxiglucosa (18F-FDG) es un análogo de la glucosa y un marcador sustituto, para el metabolismo de la glucosa. Por lo tanto, el aumento de la glucólisis en ciertas zonas, en comparación con el tejido sano, es una distintiva característica de la transformación maligna. El aumento de la glucólisis puede ser capturado utilizando PET con técnica FDG, que permite la diferenciación entre tejido normal y maligno. El híbrido de FDG-PET y los datos morfológicos de la TC tienen potencial, para mejorar la especificidad del PET.
Varios estudios realizados con 18 FDG-PET y PET-TC, apoyaron la eficacia en la detección de metástasis ganglionar loco regional, a distancia y posteriormente, la estadificación en pacientes con CCM. 20
El inconveniente más importante de esta técnica, es que en algunos casos el atrapamiento metabólico puede ser inespecífico y además de las células tumorales, también se puede encontrar en sitios de inflamación o infección. En el caso de metástasis cerebrales, FDG-PET se ve obstaculizada debido a la alta tasa metabólica cerebral. 16,18,20
PET-TC puede obtener imagen de todo el cuerpo en un solo estudio.
DIAGNÓSTICOS DIFERENCIALES
El CCM puede presentar gran similitud macroscópica e histológica con otras lesiones, por lo que el estudio histopatológico e inmunohistoquímico, permiten realizar el diagnóstico diferencial.
Se incluyen dentro de los diagnósticos diferenciales (algunos ya mencionados anteriormente):
- Melanoma amelanótico y pigmentado.
- Linfomas cutáneos.
- Metástasis de cáncer de pulmón a células pequeñas.
- Metástasis a piel de cualquier origen.
- Carcinoma epidermoide pobremente diferenciado.
- Carcinoma basocelular.
- Queratoacantoma.
- Rabdomiosarcoma.
- Neuroblastoma del adulto.
- Dermatofibrosarcoma protuberans.
- Osteosarcoma de células pequeñas.
- Sarcoma de Ewing extraesquelético. 2,3,4,8,9,12
Los patrones inmunohistoquímicos útiles para diferenciarlos del CCM son: en el caso del rabdomiosarcoma se tiñe con colorantes de actina, desmina, mioD y miogenina específicas de músculo; en el dermatofibrosarcoma protuberans, en la mayoría de los casos (50-90%), se expresa el antígeno CD34, entre otros marcadores menos específicos y generalmente son negativos para el factor XIIIa, S-100, HMB 45, CK 20, AE1/AE2, marcadores neuroendócrinos y otros; en el neuroblastoma del adulto se observa inmunorreacción positiva para sinaptofisina, cromogranina, enolasa, NB84 (anticuerpo monoclonal), CD 56 y S-100; en los casos de sarcoma de Ewing extraesquelético y osteosarcoma de células pequeñas, se destaca el anticuerpo monoclonal CD 99. 14,21
ESTADIFICACIÓN Y PRONÓSTICO
Según el American Joint Committe on Cancer existen cuatro etapas clínicas en el CCM, de acuerdo con las características en el momento de la presentación. 9
Estadios:
I. Enfermedad localizada: lesión primaria igual o menor a 2 cm.
II. Enfermedad localizada: lesión primaria mayor a 2 cm.
III. Invasión ganglionar (es el caso de nuestra paciente).
IV. Enfermedad metastásica más allá de los ganglios locales (hígado, hueso, sistema nervioso central, pulmón, páncreas, piel, glándula suprarrenal).
La sobrevida después del diagnóstico de CCM depende en gran medida del estadio de presentación. Los pacientes con verdadera enfermedad local (estadio I y II), tienen más del 90% de probabilidades de sobrevivir a los cinco años, porcentaje que decrece en aproximadamente el 60%, cuando están afectados los ganglios (estadio III) y a menos del 10% entre los que presentan enfermedad metastásica (estadio IV). A diferencia del melanoma, si el CCM recurre tiende a hacerlo muy rápido, detectándose el 90% de las recurrencias en los tres años posteriores al diagnóstico. 9,12
Como se ha señalado previamente, se encuentra descrita, otra vez al igual que en el melanoma, la regresión espontánea del tumor, en forma de regresión espontánea completa primaria (después de una biopsia incisional) y después de la recurrencia local o regional de la neoplasia (regresión espontánea completa secundaria). 10,12,22
Hasta en un 10-20% de los casos se presentan sin tumor primario conocido, lo que parece encontrarse asociado a un mejor pronóstico. 10,12
Según la bibliografía consultada 2,3,4,6,7,10,12 son considerados indicadores de mal pronóstico:
- Varones.
- Segundas neoplasias.
- Tratamiento inmunosupresor previo.
- Recidiva local.
- Edad avanzada.
- Localización en tronco.
- Tamaño tumoral mayor a 2 cm.
- Invasión linfática o vascular.
- Patrón de crecimiento difuso en la histología.
- Más de 10 mitosis por campo en la histopatología (Ki-67 elevado).
A partir del quinto “ítem” en adelante, se encuentran presentes estas características en el caso que presentamos.
TRATAMIENTO
La terapia óptima para el CCM es un tema controvertido y no existe consenso, acerca de la terapéutica más adecuada para el CCM en los estadios iniciales y en particular, sobre el tratamiento adyuvante postquirúrgico. El uso de quimioterapia en recurrencias y enfermedad metastásica, se complica por la elevada edad de los pacientes, que tienden a tolerar peor el tratamiento. No obstante, puede resumirse, básicamente, en la siguiente:
Resección quirúrgica en el sitio primario: es el tratamiento de elección. Debido a que este tumor muestra un crecimiento vertical intenso, deben examinarse los márgenes profundos cuidadosamente: márgenes sanos amplios de 2-3 cm aproximadamente y hasta la fascia muscular en profundidad, sin afectarla, aunque los márgenes se irán reduciendo seguramente.
La escisión quirúrgica, puede ser el único tratamiento o combinarse con radioterapia adyuvante del lecho quirúrgico y ganglios de drenaje, lo que disminuye la tasa de recurrencia.
Si el tumor se localiza en el pabellón auricular, algunos autores recomiendan realizar una parotidectomía superficial. 9,12,17,23
Cirugía de Mohs: la cirugía micrográfica de Mohs ha demostrado ventajas, frente a la extirpación amplia en algunas series. Demostró disminuir la tasa de recurrencia local, por lo que sería de gran utilidad especialmente en aquellos casos que se requiere un buen resultado cosmético, y algunos autores, refieren que se permite un mejor control loco-regional que con la cirugía tradicional, debido a la evaluación histológica del 100% de los márgenes quirúrgicos, incluyendo el margen profundo. Este último, es importante por la tendencia de estas neoplasias a mostrar un extenso crecimiento vertical, por lo que el margen profundo puede encontrarse afecto después de la cirugía.
El empleo de radioterapia después de la cirugía de Mohs, puede contribuir a reducir el riesgo de recidiva loco-regional, sobre todo en tumores grandes o recurrentes. 9,12,23
Linfadenectomía regional: la linfadenectomía regional terapéutica está indicada en aquellos pacientes con afectación ganglionar clínica o radiológica. Resulta controvertido, sin embargo, el empleo de radioterapia o disección ganglionar profiláctica, aunque la afectación loco-regional después de la resección quirúrgica aislada se aproxima al 50%, algunos autores consideran que todos los pacientes deberían considerarse de alto riesgo e indicar dicha linfadenectomía.17 Otros recomiendan el empleo de linfadenectomía profiláctica en aquellos casos localizados en cabeza y cuello, con diámetro tumoral >1.5 cm o evidencia histológica de invasión vascular o linfática, a pesar que no existen datos fehacientes acerca de la mejoría de los pacientes, incluidos en estos subgrupos con esta terapéutica. 23
Actualmente, se aconseja realizar linfadenectomía regional sólo si se detectan adenopatías palpables y si estas no se detectan clínicamente, realizar biopsia del ganglio centinela ya que, al igual que en el melanoma, la práctica de la linfadenectomía profiláctica no ha demostrado un incremento de la supervivencia y sí de la morbilidad. 9,12,17,23
También se considera necesaria una parotidectomía, cuando el tumor utiliza como vía linfática de drenaje los linfáticos parotídeos. 12
Radioterapia: debido a que el CCM es radiosensible, la radioterapia sigue siendo una alternativa terapéutica útil para el control de la recurrencia local.
El uso de la radioterapia incluye:
- Tratamiento postoperatorio (adyuvante) del lecho quirúrgico y de los ganglios linfáticos regionales, con dosis entre 45-50 Gy para sitio primario con márgenes de resección negativos y lecho ganglionar sin enfermedad palpable. Dosis de 55-60 Gy para sitio primario con márgenes de resección positivos y lecho ganglionar con enfermedad palpable. Se recomienda administrarlas en 20 – 25 sesiones durante unas cuatro a seis semanas. Se ha descrito una disminución del riesgo local de recidivas con el uso de radioterapia adyuvante, tras el tratamiento quirúrgico (10,5 % frente a 52,6%) y un aumento de la supervivencia libre de enfermedad (88 frente a 58 meses). Sin embargo, esta terapia combinada no ha demostrado un aumento significativo de la supervivencia global. 9,24,25,26
- Tratamiento primario para tumores irresecables o pacientes inoperables, como tratamiento paliativo. 24,25
- Radioterapia de rescate para enfermedad recurrente. 24,25,26
Los efectos adversos agudos de la radioterapia son el eritema local y la fatiga leve a moderada, que se acentúa hacia el final de la terapia y en general desaparece en uno a dos meses, después de finalizado un tratamiento de cinco semanas.
Los cambios crónicos en la piel debido a la radiación incluyen: alopecia permanente o temporaria en el lugar irradiado, atrofia de la epidermis, pérdida de los anexos que conlleva sequedad de la piel o las mucosas y riesgo de cáncer de piel secundario en la región tratada, en pacientes con una expectativa de vida superior a los 20 años. Tal vez el efecto colateral potencial más importante es el linfedema. Esto representa un problema más frecuente en los miembros inferiores, cuando la radioterapia se aplica en los ganglios inguinales, especialmente después que se ha realizado una cirugía en la misma región, como es el caso de nuestra paciente. 9,24
Quimioterapia: la quimioterapia no está indicada como primera elección, su empleo, aunque poco alentador, puede ser considerado en las siguientes circunstancias:
- Tratamiento postoperatorio (adyuvante): la quimioterapia adyuvante no parece disminuir los índices de recurrencia ni mejorar la supervivencia.
- Tratamiento de la enfermedad de alto riesgo, asociada a radioterapia.
- Tratamiento de las metástasis a distancia: los agentes quimioterápicos habituales usados para CCM en estadios avanzados, son los mismos que los usados para el carcinoma de células pequeñas de pulmón. La asociación con radioterapia parece que ofrece resultados más favorables. 9,26,27
La poliquimioterapia (cisplatino, carboplatino, etopósido, doxorrubicina, entre otras) es útil pero aún no hay un esquema establecido. 27
Otros: otras modalidades terapéuticas descritas en la bibliografía son: la hipertermia local, la administración intralesional directa de factor de necrosis tumoral humano recombinante y la electroquimioterapia. 9,27,28,29 Faltan más estudios que avalen estas técnicas.
El mejor resultado se obtiene claramente cuando se desarrolla un tratamiento multidisciplinario, llevado a cabo por un equipo con experiencia. 30
EVOLUCIÓN Y CONTROL EVOLUTIVO
Después del tratamiento de las lesiones primarias del CCM, se requiere una vigilancia estrecha dado el alto índice de recidivas y resulta de gran importancia, el seguimiento del paciente mediante la exploración física, analítica e imagenológica. Algunos autores recomiendan revisiones cada mes durante los primeros seis meses, cada dos o tres meses durante los dos años siguientes y posteriormente, cada seis meses.
Control por imágenes en el carcinoma de células de Merkel: según la bibliografía consultada, para imágenes de seguimiento, se sugiere una radiografía de tórax de rutina, así como una tomografía computarizada de la región afectada, tres meses después de la terapia. Cada año después de la terapia se recomienda una radiografía de tórax, TC y la resonancia magnética. 16,18
Debido al bajo costo de la ecografía, tiene un alto valor en la rutina de imágenes de seguimiento en el CCM. La radiografía de tórax es una técnica de imagen de rutina, para evaluar la posible afectación pulmonar.
Peloschek y col recomiendan la repetición de la FDG-PET a los tres meses y un año después del tratamiento.16
CONCLUSIONES
En la paciente presentada, se destaca la presencia de carcinoma de células de Merkel con una localización atípica y de gran tamaño, lo que es poco frecuente según los casos publicados en la bibliografía consultada. Además es necesario enfatizar en que no nos debemos olvidar de incluir al CCM en los diagnósticos diferenciales, ya que en el 90% de los casos, incluida nuestra paciente, se lo diagnostica por el resultado del examen histopatológico y aunque la mayoría de los pacientes y médicos no están familiarizados con la enfermedad o su tratamiento específico, puede ser mortal y la terapia debe iniciarse con rapidez.
También debemos recordar que la inmunohistoquímica es característica para el diagnóstico, aunque no exclusiva.
Con la información limitada de estudios prospectivos que nos guíen, las estrategias de manejo óptimo para CCM aún no se han definido claramente. Sin embargo, nuestra comprensión del CCM está mejorando, poco a poco y las estrategias más eficaces están saliendo lentamente de la incertidumbre. En lo que se refiere a la biopsia del ganglio centinela en pacientes con CCM, son necesarios más diseños metodológicos y estudios prospectivos para aclarar su papel.
Respecto del tratamiento, el mejor resultado se obtiene claramente cuando se desarrolla un tratamiento multidisciplinario, llevado a cabo por un equipo con experiencia.
AGRADECIMIENTOS
A la paciente en primer lugar. A los Dres. Ricardo Achenbach, Eduardo Píttaro y Mónica Dutto, por guiarme en la elaboración de este trabajo y en la búsqueda bibliográfica y a la Asociación Argentina de Dermatología, por servirme de apoyo para el buen desempeño del mismo.
1. Sánchez G. Serotonina (5-HT) y catecolaminas en la célula de Merkel. Arch Argent Dermatol 2007; 57: 175-183.
2. Díaz Mathe A, Vargas A, González VM, Casas J, Larralde M. Carcinoma de células de Merkel. Rev Argent Dermatol 2009; 15 (2): 134-136.
3. Saadi ME, Alarcón B, Abeldaño A, Brea P, Kien MC, Chouela E. Carcinoma de células de Merkel. Rev Argent Dermatol 2002; 8 (2): 218-222.
4. Cañadas NG, Luna PC, Nocito MJ, Lustia MM, Etcheverry ML, Castellanos Posse ML, Marchesi C, Garuti RA, Carabajal G, Mazzini MA. Carcinoma de células de Merkel. Estudio de 5 casos. Rev Argent Dermatol 2009; 15 (6): 428-433.
5. Magnin PH, Casas JG, Farjat ME. Evolución del carcinoma trabecular. Rev Argent Dermatol 1986; 67: 333-340.
6. Youlden DR, Soyer HP, Youl PH, Fritschi L, Baade PD. Incidence and survival for Merkel cell carcinoma in Queensland. Australia, 1993-2010. JAMA Dermatol 2014; 150 (8): 864-872.
7. Girschik J, Thorn K, Beer TW. Merkel cell carcinoma in Western Australia: a population-based study of incidence and survival. Br J Dermatol 2011; 165: 1051-1057.
8. Gómez Sánchez ME, Martínez Martínez ML, Berruga CF, López Villaescusa MT, Rodríguez Vázquez M, Pérez García LP. Carcinoma de células de Merkel. A propósito de un caso. Rev Clin Med Fam 2013; 6 (1): 43-46.
9. Nghiem P, Jaimes N. Carcinoma de células de Merkel. En: Wolff K, Goldsmith L, Katz S, Gilchrest B, Paller A, Leffell D. Dermatología en Medicina General de Fitzpatrick. Séptima Edición. Tomo 2. Editorial Médica Panamericana. Buenos Aires. Argentina. 2009; 1087-1094.
10. Albores-Saavedra J, Batich K, Chable-Montero F. Merkel cell carcinoma demographics, morphology, and survival base on 3870 cases: a population based study. J Cutan Pathol 2010; 37: 20-27.
11. Shuda M, Chang Y, Moore P. Merkel cell polyomavirus positive Merkel cell carcinoma requires viral small T antigen for cell proliferation. J Invest Dermatol 2014; 134 (5): 1479-1481.
12. Pulitzer MP, Amin BD, Busam KJ. Merkel cell carcinoma: review. Adv Anat Pathol 2009; 16: 135-144.
13. Llombart B, Monteagudo C, López-Guerrero JA, Carda C y col. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005; 46: 622-634.
14. Fuertes L, Santonja C, Kutzner H, Requena L. Inmunohistoquímica en dermatopatología: revisión de los anticuerpos utilizados con mayor frecuencia (parte II). Actas Dermosifiliogr 2013; 104 (3): 181-203.
15. Henderson SA, Tetzlaff MT, Pattanaprichakul P, Fox P y col. Detection of mitotic figures and G2+ tumor nuclei with histone markers correlates with worse overall survival in patients with Merkel cell carcinoma. J Cutan Pathol 2014; 41: 846-852.
16. Enzenhofer E, Ubl P, Czerny C, Erovic BM. Imaging in Patients with Merkel Cell Carcinoma. J Skin Cancer 2013; 2013: 1-6.
17. Asgari MM, Sokil MM, Warton EM, Iyer J, Paulson KG, Nghiem P. Effect of host, tumor, diagnostic and treatment variables on outcomes in a large cohort with Merkel cell carcinoma. JAMA Dermatol 2014; 150 (7): 716-723.
18. Peloschek P, Novotny C, Mueller-Mang C. Diagnostic imaging in Merkel cell carcinoma: lessons to learn from16 cases with correlation of sonography, CT, MRI and PET. Eur J Radiol 2010; 73 (2): 317-323.
19. Thompson JF, Hruby G. The role of sentinel lymph node biopsy in patients with Merkel cell carcinoma: uncertainty prevails. Ann Surg Oncol 2014; 21: 1517-1519.
20. Concannon R, Larcos GS, Veness M. The impact of 18 F-FDG PET-CT scanning for staging and management of Merkel cell carcinoma: Results from Westmead Hospital, Sydney, Australia. J Am Acad Dermatol 2010; 62 (1): 76-84.
21. Pérez OG, Solarz H, Amante H, Woscoff A. Dermatofibrosarcoma protuberans: actualización inmunohistoquímica. Dermatol Argent 2008; 14 (3): 220-224.
22. Bertolotti A, Conte H, Francois L, Dutriaux C y col. Merkel cell carcinoma: complete clinical remission associated with disease progression. JAMA Dermatol 2013; 149 (4): 501-502.
23. Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg 2013; 2013: 1-17.
24. Kim JA, Choi AH. Effect of radiation therapy on survival in patients with resected Merkel cell carcinoma. A propensity score surveillance, epidemiology and end results database analysis. JAMA Dermatol 2013; 149 (7): 831-838.
25. Hruby G, Scolyer RA, Thompson JF. The important role of radiation treatment in the management of Merkel cell carcinoma. Br J Dermatol 2013; 169: 975-982.
26. Lewis KG, Weinstock MA, Weaver AL, Otley CC. Adjuvant Local Irradiation for Merkel cell carcinoma. Arch Dermatol 2006; 142: 693-700.
27. Desch L, Kunstfeld R. Merkel cell carcinoma: chemotherapy and emerging new therapeutic options. J Skin Cancer 2013; 2013: 1-9.
28. Miller NJ, Bhatia S, Parvathaneni U, Iyer JG y col. Emerging and mechanism-based therapies for recurrent or metastasic Merkel cell carcinoma. Curr Treat Options Oncol 2013; 14 (2): 249-263.
29. Curatolo P, Mancini M, Clerico R, Ruggiero A y col. Remission of extensive Merkel cell carcinoma after electrochemotherapy. Arch Dermatol 2009; 145 (4): 494-495.
30. Miranda S, Gbaguidi X, Carvalho P, Chassagne J. Merkel cell carcinoma: the impact of multidisciplinary management. J Nutr Health Aging 2013; 17 (2): 196-198.
Referencias
REFERENCIAS
1. Sánchez G. Serotonina (5-HT) y catecolaminas en la célula de Merkel. Arch Argent Dermatol 2007; 57: 175-183.
2. Díaz Mathe A, Vargas A, González VM, Casas J, Larralde M. Carcinoma de células de Merkel. Rev Argent Dermatol 2009; 15 (2): 134-136.
3. Saadi ME, Alarcón B, Abeldaño A, Brea P, Kien MC, Chouela E. Carcinoma de células de Merkel. Rev Argent Dermatol 2002; 8 (2): 218-222.
4. Cañadas NG, Luna PC, Nocito MJ, Lustia MM, Etcheverry ML, Castellanos Posse ML, Marchesi C, Garuti RA, Carabajal G, Mazzini MA. Carcinoma de células de Merkel. Estudio de 5 casos. Rev Argent Dermatol 2009; 15 (6): 428-433.
5. Magnin PH, Casas JG, Farjat ME. Evolución del carcinoma trabecular. Rev Argent Dermatol 1986; 67: 333-340.
6. Youlden DR, Soyer HP, Youl PH, Fritschi L, Baade PD. Incidence and survival for Merkel cell carcinoma in Queensland. Australia, 1993-2010. JAMA Dermatol 2014; 150 (8): 864-872.
7. Girschik J, Thorn K, Beer TW. Merkel cell carcinoma in Western Australia: a population-based study of incidence and survival. Br J Dermatol 2011; 165: 1051-1057.
8. Gómez Sánchez ME, Martínez Martínez ML, Berruga CF, López Villaescusa MT, Rodríguez Vázquez M, Pérez García LP. Carcinoma de células de Merkel. A propósito de un caso. Rev Clin Med Fam 2013; 6 (1): 43-46.
9. Nghiem P, Jaimes N. Carcinoma de células de Merkel. En: Wolff K, Goldsmith L, Katz S, Gilchrest B, Paller A, Leffell D. Dermatología en Medicina General de Fitzpatrick. Séptima Edición. Tomo 2. Editorial Médica Panamericana. Buenos Aires. Argentina. 2009; 1087-1094.
10. Albores-Saavedra J, Batich K, Chable-Montero F. Merkel cell carcinoma demographics, morphology, and survival base on 3870 cases: a population based study. J Cutan Pathol 2010; 37: 20-27.
11. Shuda M, Chang Y, Moore P. Merkel cell polyomavirus positive Merkel cell carcinoma requires viral small T antigen for cell proliferation. J Invest Dermatol 2014; 134 (5): 1479-1481.
12. Pulitzer MP, Amin BD, Busam KJ. Merkel cell carcinoma: review. Adv Anat Pathol 2009; 16: 135-144.
13. Llombart B, Monteagudo C, López-Guerrero JA, Carda C y col. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005; 46: 622-634.
14. Fuertes L, Santonja C, Kutzner H, Requena L. Inmunohistoquímica en dermatopatología: revisión de los anticuerpos utilizados con mayor frecuencia (parte II). Actas Dermosifiliogr 2013; 104 (3): 181-203.
15. Henderson SA, Tetzlaff MT, Pattanaprichakul P, Fox P y col. Detection of mitotic figures and G2+ tumor nuclei with histone markers correlates with worse overall survival in patients with Merkel cell carcinoma. J Cutan Pathol 2014; 41: 846-852.
16. Enzenhofer E, Ubl P, Czerny C, Erovic BM. Imaging in Patients with Merkel Cell Carcinoma. J Skin Cancer 2013; 2013: 1-6.
17. Asgari MM, Sokil MM, Warton EM, Iyer J, Paulson KG, Nghiem P. Effect of host, tumor, diagnostic and treatment variables on outcomes in a large cohort with Merkel cell carcinoma. JAMA Dermatol 2014; 150 (7): 716-723.
18. Peloschek P, Novotny C, Mueller-Mang C. Diagnostic imaging in Merkel cell carcinoma: lessons to learn from16 cases with correlation of sonography, CT, MRI and PET. Eur J Radiol 2010; 73 (2): 317-323.
19. Thompson JF, Hruby G. The role of sentinel lymph node biopsy in patients with Merkel cell carcinoma: uncertainty prevails. Ann Surg Oncol 2014; 21: 1517-1519.
20. Concannon R, Larcos GS, Veness M. The impact of 18 F-FDG PET-CT scanning for staging and management of Merkel cell carcinoma: Results from Westmead Hospital, Sydney, Australia. J Am Acad Dermatol 2010; 62 (1): 76-84.
21. Pérez OG, Solarz H, Amante H, Woscoff A. Dermatofibrosarcoma protuberans: actualización inmunohistoquímica. Dermatol Argent 2008; 14 (3): 220-224.
22. Bertolotti A, Conte H, Francois L, Dutriaux C y col. Merkel cell carcinoma: complete clinical remission associated with disease progression. JAMA Dermatol 2013; 149 (4): 501-502.
23. Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg 2013; 2013: 1-17.
24. Kim JA, Choi AH. Effect of radiation therapy on survival in patients with resected Merkel cell carcinoma. A propensity score surveillance, epidemiology and end results database analysis. JAMA Dermatol 2013; 149 (7): 831-838.
25. Hruby G, Scolyer RA, Thompson JF. The important role of radiation treatment in the management of Merkel cell carcinoma. Br J Dermatol 2013; 169: 975-982.
26. Lewis KG, Weinstock MA, Weaver AL, Otley CC. Adjuvant Local Irradiation for Merkel cell carcinoma. Arch Dermatol 2006; 142: 693-700.
27. Desch L, Kunstfeld R. Merkel cell carcinoma: chemotherapy and emerging new therapeutic options. J Skin Cancer 2013; 2013: 1-9.
28. Miller NJ, Bhatia S, Parvathaneni U, Iyer JG y col. Emerging and mechanism-based therapies for recurrent or metastasic Merkel cell carcinoma. Curr Treat Options Oncol 2013; 14 (2): 249-263.
29. Curatolo P, Mancini M, Clerico R, Ruggiero A y col. Remission of extensive Merkel cell carcinoma after electrochemotherapy. Arch Dermatol 2009; 145 (4): 494-495.
30. Miranda S, Gbaguidi X, Carvalho P, Chassagne J. Merkel cell carcinoma: the impact of multidisciplinary management. J Nutr Health Aging 2013; 17 (2): 196-198.


