Autores | Contacto
V Chiesura *, M Vásquez *, E Valente **, M Kurpis *** y A Ruiz Lascano ****
* Médica Residente 2º Año de Dermatología. Hospital Privado de Córdoba. Alumna de Postgrado de la Carrera en Dermatología. Universidad Católica de Córdoba.
* Médica Residente 1º Año de Dermatología. Hospital Privado de Córdoba. Alumna de Postgrado de la Carrera en Dermatología. Universidad Católica de Córdoba.
**Médico de Planta del Servicio de Dermatología. Hospital Privado de Córdoba.
*** Médica de Planta del Servicio de Anatomía Patológica. Hospital Privado de Córdoba.
****Jefe del Servicio de Dermatología. Hospital Privado de Córdoba. Director del Postgrado de la Carrera en Dermatología. Universidad Católica de Córdoba.
Servicio de Dermatología Hospital Privado de Córdoba. Carrera de Postgrado en Dermatología. Universidad Católica de Córdoba.
Autor Responsable: Vilma Carolina Chiesura
Dirección Postal: Naciones Unidas 346 (X5016KEH) Córdoba. Argentina. Teléfono: +54-351-4688810
E-mail: vilmachiesura@gmail.com
Recibido: 11-06-2014
Aceptado para su publicación: 31-08-2014
Dirección
Prof. Dr. Ricardo E. Achenbach
Resumen | Palabras Claves
RESUMEN
Presentamos el caso de una mujer de 71 años de edad, que consulta en el Servicio de Dermatología del Hospital por placas eritematosas, edematosas, induradas al tacto en antebrazos y piernas, que progresan con franca induración del tegumento, limitación de la movilidad articular, contracturas en flexión y debilidad muscular leve, asociado a placas esclerodermiformes en tronco. La histopatología informó epidermis y dermis conservada con fascitis eosinofílica evolucionada asociada a miositis. Se revisaron las formas de presentación clínica, los métodos diagnósticos (histopatología y resonancia magnética), diagnósticos diferenciales y posibles tratamientos.
PALABRAS CLAVE: Fascitis eosinofílica; Miositis.
SUMMARY
We report the case of a 71 years old woman, consulting in the Dermatology Department of our hospital for erythematous, edematous, indurated to the touch patches in forearms and legs, progressing to frank induration of the integument, limited joint mobility, flexion contractures and mild muscle weakness associated with scleroderma-like plaques on the trunk. The histopathology reported epidermis and dermis preserved with eosinophilic fasciitis and myositis. The clinical presentation, diagnostic methods (histopathology and MRI), differential diagnosis and possible treatments, were reviewed.
KEY WORDS: Eosinophilic fasciitis; Myositis.
Artículo | Referencias
Descargar archivo PDF aquí
INTRODUCCIÓN
La fascitis eosinofílica (FE) o síndrome de Shulman es una enfermedad poco común, de etiología desconocida, que se presenta entre los 20-60 años de edad, en ambos sexos por igual 1,2,3,4.
Clínicamente se caracteriza por una induración leñosa, simétrica, de la piel y tejidos blandos de miembros superiores (88%), miembros inferiores (70 %), cuello (6-18 %) y tronco (17-32 %). Suele respetar la cara, los dedos de las manos y los pies y no afecta vísceras 1,4.
El comienzo de la enfermedad es usualmente agudo y puede acompañarse de pérdida de peso, astenia y mialgias espontáneas o provocadas 1,4.
El diagnóstico de FE se basa generalmente en la asociación de las alteraciones de la piel y los tejidos blandos, con un infiltrado inflamatorio compuesto en su mayoría por linfocitos y eosinófilos. Sin embargo, aunque la presencia de eosinofilia periférica es frecuente, no es indispensable para el diagnóstico de FE 4.
Múltiples causas han sido identificadas como posibles responsables de esta patología: ejercicio extremo, trauma, iniciación de hemodiálisis, infección por Borrellia burgdorferi, radioterapia, drogas (atorvastatina, simvastatina, ramipril, fenitoina, heparina, ibuprofeno, cimetidina, hidroxicloroquina), entre otras 1-4. Sin embargo, la mayoría de los casos no están asociados con estos factores y son considerados idiopáticos 1.
Presentamos un caso de FE avanzada asociada a miositis y revisamos la literatura, acerca de su forma de presentación clínica, diagnósticos diferenciales, métodos diagnósticos (histopatología y resonancia magnética -RMN-) y posibles tratamientos.
CASO CLÍNICO
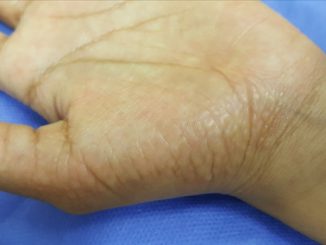
Mujer de 71 años de edad, con antecedentes de hipertensión arterial, hipotiroidismo, prolapso de válvula mitral e insuficiencia aórtica leve, arritmias y síndrome Sicca, polimedicada. Consulta a mediados de 2011 en el Servicio de Dermatología, por presentar placas eritematosas, edematosas, induradas al tacto, de dos meses de evolución en antebrazos y piernas (Fig 1). No presentaba alteración de la apertura bucal, esclerodactilia o debilidad muscular; negaba síntomas gastrointestinales, renales o fenómeno de Raynaud. El laboratorio mostraba 12 % de eosinófilos y TSH elevada. Se realizó una TAC de tórax, abdomen y pelvis dentro de parámetros normales. La histopatología informó dermatitis perivascular a predominio linfocítico con edema y fibrosis dérmica y se consideró insuficiente, por no abarcar fascia ni músculo ante la sospecha clínica de FE.

Fig 1: placas eritematosas, edematosas, induradas al tacto, de dos meses de evolución en antebrazos.
Debido a que la paciente debió ser hospitalizada por un cuadro de meningoencefalitis herpética, regresó a la consulta un año después. La piel de antebrazos y piernas era móvil en superficie, pero adherida a planos profundos. La progresión franca en la induración de los tegumentos, limitaba la movilidad articular y se asociaba a debilidad muscular leve. Una nueva biopsia por losange informó: marcada fibrosis hialina con infiltrado inflamatorio, a predominio mononuclear y eosinófilos ocasionales a nivel de la fascia muscular, con extensión del infiltrado hacia las fibras musculares estriadas adyacentes, compatible con FE evolucionada asociada a miositis (Fig 2).

Fig 2: H&E, 400 X: fibrosis hialina con infiltrado inflamatorio, a predominio mononuclear y eosinófilos ocasionales, a nivel de la fascia muscular (flecha roja superior), con extensión del infiltrado hacia las fibras musculares estriadas adyacentes (flecha roja inferior), compatible con FE evolucionada asociada a miositis.
En los estudios laboratoriales presentaba CPK, aldolasa y colagenograma normal. En valoración conjunta con el Servicio de Reumatología, se inició tratamiento con prednisona 40 mg/día y metotrexato 20 mg/semanales.
Tres meses después se decidió la internación de la paciente para valoración y tratamiento, debido a la progresión clínica de la enfermedad. Además de la franca induración de piernas y antebrazos, presentaba placas blanquecinas, esclerodermiformes, en región dorsolumbar en espalda y región lateral derecha del tórax (Fig 3). En el tercio superior de los muslos, la piel presentaba aspecto edematoso tipo “piel de naranja” (Fig 4).

Fig 3: placas blanquecinas, esclerodermiformes, en región dorsolumbar en espalda y región lateral derecha del tórax (flechas rojas).

Fig 4: piel de aspecto edematoso tipo “piel de naranja”, en el tercio superior del muslo izquierdo (flecha roja).
El laboratorio mostraba plaquetopenia, VSG elevada, LDH elevada, pico monoclonal gamma en el proteinograma por electroforesis, con IgM sérica elevada e inmunofijación en orina con proteinuria kappa. La punción aspiración de médula ósea informó: médula ósea hipercelular con dismegacariopoyesis. Los valores de CPK y el colagenograma fueron normales nuevamente.
Una resonancia magnética nuclear de cintura escapular, pelvis y muslos informó: aumento de la intensidad de señal en T2, a nivel de la fascia de los músculos glúteos (Fig 5). La capilaroscopía fue considerada no diagnóstica (capilares finos y elongados con zonas avasculares).

Fig 5: RMN: pelvis y muslos: se evidencia aumento de intensidad de señal en T2 a nivel de la fascia de los músculos glúteos (flechas rojas).
Debido a la falta de respuesta terapéutica, se realizó una nueva biopsia que reafirmó los resultados previos: epidermis y dermis conservada, tejido celular subcutáneo con ligera fibrosis septal, marcada fibrosis hialina e infiltrado inflamatorio mononuclear, con eosinófilos ocasionales a nivel de la fascia muscular, con progresión del infiltrado a las fibras musculares estriadas adyacentes, que se observaron atróficas en relación a la muestra previa (Figs 6 y 7).

Fig 6: H&E, 400 X: se observa marcada fibrosis hialina e infiltrado inflamatorio mononuclear, con eosinófilos ocasionales a nivel de la fascia muscular.

Fig 7: H&E, 400 X: progresión del infiltrado a las fibras musculares estriadas adyacentes, que se observaron atróficas en relación a la muestra previa.
La paciente fue tratada con pulsos de metilprednisolona 500 mg/día por tres días. Progresó con insuficiencia ventilatoria restrictiva y cuadro de urosepsis, constatándose el óbito un mes después.
DISCUSIÓN
La fascitis eosinofílica (FE) es una patología rara, que afecta la piel y los tejidos blandos de los miembros y el tronco 4.
La etapa inicial de la enfermedad cursa inicialmente con eritema y edema en las extremidades, que luego progresa a induración simétrica. La resolución del edema otorga al tegumento el aspecto de “piel de naranja”. En la etapa tardía predomina el engrosamiento del colágeno de la dermis, tejido celular subcutáneo (TCS) y fascia con una textura irregular y leñosa, que difiere de la piel de superficie suave y brillante, que se observa en pacientes con esclerodermia 1.
El signo de Groove es un hallazgo físico de esta patología, caracterizado por la indentación visible de las venas superficiales al elevar los miembros afectados. Esto reduce la presión venosa y evidencia el respeto relativo de la epidermis y las capas superficiales de la dermis, en contraste con el proceso de fibrosis de las capas más profundas adyacentes a las venas 1-4.
La enfermedad puede cursar con una serie de manifestaciones clínicas extracutáneas, tales como: 1) artritis, limitación de movilidad articular y contracturas en flexión; 2) neuropatías periféricas y craneales (síndrome del túnel carpiano, mononeuritis múltiple); 3) mialgias leves y debilidad muscular (raro miositis inflamatoria) 1. La paciente del caso presentaba las manifestaciones osteoarticulares y musculares descritas.
En general, no se ha descrito compromiso de órganos internos por el proceso fibrótico 1. Algunos autores citan ciertos ejemplos de compromiso visceral tales como: dismotilidad esofágica, defectos de difusión y restricción pulmonar, derrame pericárdico y alteraciones renales, entre las manifestaciones extracutáneas de la enfermedad. Sin embargo, estas manifestaciones sólo han sido comunicadas en unos pocos casos y probablemente correspondan a esclerodermias sistémicas 3,4. La paciente presentó una insuficiencia ventilatoria restrictiva coincidiendo con la literatura.
La FE ha sido asociada a: 1) trastornos endocrinológicos: tiroiditis, hipotiroidismo; 2) trastornos hematológicos: anemia aplásica, trombocitopenia amegacariocítica adquirida, desórdenes mieloproliferativos, síndromes mielodisplásicos, leucemia, linfoma y mieloma múltiple 1,4; 3) neoplasias malignas de órganos sólidos: cáncer de mama, melanoma de coroides, cáncer broncopulmonar, cáncer de próstata 3,4. Debido a la escasez de casos relacionados a tumores de órganos sólidos, la necesidad de evaluar una neoplasia subyacente en pacientes con FE está en discusión, al menos que exista alguna manifestación clínica sugestiva 4.
El presente caso podría asociarse a trastornos hematológicos, ya que, la paciente presentaba una gammapatía monoclonal de significado desconocido, así como a trastornos endocrinológicos, debido a que tenía antecedentes personales de hipotiroidismo.
El hallazgo de laboratorio más frecuente es la eosinofilia periférica (63-93%), que suele ser transitoria y encontrarse solamente en la fase aguda de la enfermedad. Otros hallazgos son: PCR elevada, VSG elevada (29-36%) e hipergammaglobulinemia policlonal (35-50 %). Los niveles de CPK pueden ser normales, incluso en pacientes con mialgias (raramente elevado en el 4 a 6 % de los casos). Los ANA, en general negativos, pueden detectarse en el 15 a 20 % de los pacientes. Se espera que ANCA, ENA y anti DNA sean negativos 1,3,4.
La maniobra fundamental para el diagnóstico, es la realización de una biopsia incisional que abarque piel, fascia y músculo de la región afectada. En la etapa temprana de la enfermedad se observa: edema e infiltrado de linfocitos, células plasmáticas, histiocitos y eosinófilos a nivel del tejido celular subcutáneo y la fascia muscular. En la etapa tardía, la dermis profunda, el tejido celular subcutáneo y la fascia se encuentran engrosados y escleróticos. En ocasiones la histopatología puede evidenciar engrosamiento e inflamación de epimisio, perimisio y endomisio y en menor grado de fibras musculares, que hacen a esta patología difícil de diferenciar de otras miopatías inflamatorias 1.
La IRM puede resultar de valor en casos atípicos de fascitis. También es útil para guiar la biopsia de piel y para monitorizar la respuesta terapéutica. El incremento de la señal en T2 en el tejido celular subcutáneo y la fascia y el realce de dichas estructuras, después de la administración de gadolinio en T1, son algunos de los hallazgos sugestivos de esta patología 1,4,5,6,7.
El presente caso, coincide con los hallazgos histopatológicos y las características de diagnóstico por imágenes comunicadas en la bibliografía; aunque la asociación es poco frecuente, la presencia de miopatía inflamatoria ha sido descrita.
La esclerodermia localizada, la esclerosis sistémica, el síndrome mialgia-eosinofilia (asociado a la ingesta de L-triptofano y 5-hidroxitriptofano) y otros desórdenes esclerodermiformes como: fibrosis nefrogénica sistémica, escleromixedema, escleredema, están incluidos entre los principales diagnósticos diferenciales de la FE 1.
Las principales características clínicas que permiten diferenciarla de la esclerodermia son: la ausencia de: esclerodactilia, fenómeno de Raynaud, alteraciones de la microcirculación objetivables por capilaroscopía y el compromiso de órganos internos 1,3. La presencia de afectación visceral, no esperable en la FE típica, debe conducirnos a excluir otras enfermedades sistémicas como síndrome de hipereosinofilia o vasculitis de Churg Strauss 4.
Para algunos autores, la FE es solo una variante clínica de la morfea, debiéndose denominar fascitis con eosinófilos (AB Ackerman) 11.
Los glucocorticoides sistémicos continúan siendo el tratamiento de primera línea para esta patología: prednisona 0.5 a 1.5 mg/kg/día o pulsos endovenosos de metilprednisolona 0.5 a 1 gr/día por días consecutivos. En pacientes con escasa respuesta a la prednisona, a dosis máxima por tres meses (1.5 mg/kg/día) pueden utilizarse otros agentes inmunosupresores como: ahorradores de corticoides (metotrexate, micofenolato de mofetilo, azatioprina, ciclosporina A, ciclofosfamida, PUVA, UVA1, infliximab, rituximab, hidroxicloroquina), etc 1,6,7,8,9. Resultados terapéuticos pobres, han sido asociados a demoras en el diagnóstico mayor a seis meses, falta de pulsos de metilprednisolona en el tratamiento inicial y la presencia de lesiones morfea-símil 10. El metotrexate a dosis de 15 a 25 mg/semana, es el tratamiento sugerido por la literatura, en pacientes con lesiones morfea-símil y escasa respuesta terapéutica a corticoides sistémicos 1,10. Los pulsos de metilprednisolona como tratamiento inicial, permitirían obtener mejores resultados terapéuticos, con menores requerimientos de inmunosupresores 10.
En lo que respecta a la duración del tratamiento de tres a seis meses con uno u otro agente, se considera tiempo suficiente para valorar la respuesta terapéutica. Una vez lograda la remisión, la terapia debe continuar entre cuatro y seis meses más, dependiendo de la respuesta clínica 1.
La fasciectomía quirúrgica se reserva para casos refractarios, síndrome de túnel carpiano y contracturas articulares 1.
El pronóstico de la FE es la resolución completa con el tratamiento habitual en el curso de uno a tres años 3,5. Ante complicaciones hematológicas o manifestaciones sistémicas, que precisan de un tratamiento inmunosupresor, el pronóstico es más desfavorable, dado que, la enfermedad evoluciona a la cronicidad y es preciso prolongar el período de tratamiento 5.
La paciente del caso presentó algunos factores de mal pronóstico para FE, como: la dificultad en el diagnóstico temprano por factores externos, alteración hematológica asociada, presencia de placas esclerodermiformes, por lo que evolucionó a la cronicidad con escasa respuesta terapéutica. La importancia del caso radica en poder observar en una misma paciente, los distintos estadios evolutivos de una patología infrecuente, que cursó con asociaciones poco comunes como la miositis y una evolución tórpida, que la llevó finalmente al óbito.
1. Helfgott S, Varga J. Eosinophilic Fasciitis. Uptodate. Sep 2012.
2. Antic M, Lautenschlager S, Itin PH. Eosinophilic Fasciitis 30 years after- What do we really know? Report of 11 Patients and Review of the Literature. Dermatology 2006; 213: 93-101.
3. Velásquez G, Gutiérrez TMA, Rosemberg GH, Figueroa EF, Bronstein AE, Jacobelli GS. Fascitis eosinofílica: experiencia de tres casos. Rev Med Chile 2002; 130 (2): 209-214.
4. Lebeaux D, Séne D. Eosinophilic Fasciitis (Shulman Disease). Best Pract Res Clin Reumatol 2012; 26: 449-458.
5. Grados Cànovas Dolors. Fascitis eosinofílica: a propósito de 13 casos. Universidad Autónoma de Barcelona 2010; 1-33.
6. Servy A, Clérici T, Malines C, Le Parc JM, Cộté JF. Eosinophilic Fasciitis: A Rare Skin Sclerosis. Pathol Res Int 2011; 1- 4.
7. Loperena Oropeza G, Sanders CE, Anderson DM, Elsheikh TM. A Case of Eosinophilic Fasciitis in a 72-Year-Old Man. Hospital Physician 2008; 35-40.
8. Paudyal BP, Gyawalee M, Sigdel K. Eosinophilic Fasciitis: A Rare Fibrosing Disorder. Kathmandu Univ Med J 2012; 39 (3): 73-75.
9. Silny W, Osmola-Mankowska A, Zarnecka-Operacz M, Żaba R, Danczak-Pazdrowska A, Marciniak A. Eosinophilic fasciitis: A report of two cases treated with ultaviolet A1 phototherapy. Photoder Photoimmunol Photomed 2009; 25: 325-327.
10. Lebeaux D, Francès C, Barete S, Wechsler B, Dubourg O, Renoux J y col. Eosinophilic Fasciitis (Shulman Disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology 2012; 51: 557-561.
11. Ackerman AB, Chongchitnant N, Sánchez J y col. Histologic Diagnosis of Inflammatory skin diseases. Segunda Edición. Williams & Wilkins. Philadelphia. EEUU. 1997.
Referencias
REFERENCIAS
1. Helfgott S, Varga J. Eosinophilic Fasciitis. Uptodate. Sep 2012.
2. Antic M, Lautenschlager S, Itin PH. Eosinophilic Fasciitis 30 years after- What do we really know? Report of 11 Patients and Review of the Literature. Dermatology 2006; 213: 93-101.
3. Velásquez G, Gutiérrez TMA, Rosemberg GH, Figueroa EF, Bronstein AE, Jacobelli GS. Fascitis eosinofílica: experiencia de tres casos. Rev Med Chile 2002; 130 (2): 209-214.
4. Lebeaux D, Séne D. Eosinophilic Fasciitis (Shulman Disease). Best Pract Res Clin Reumatol 2012; 26: 449-458.
5. Grados Cànovas Dolors. Fascitis eosinofílica: a propósito de 13 casos. Universidad Autónoma de Barcelona 2010; 1-33.
6. Servy A, Clérici T, Malines C, Le Parc JM, Cộté JF. Eosinophilic Fasciitis: A Rare Skin Sclerosis. Pathol Res Int 2011; 1- 4.
7. Loperena Oropeza G, Sanders CE, Anderson DM, Elsheikh TM. A Case of Eosinophilic Fasciitis in a 72-Year-Old Man. Hospital Physician 2008; 35-40.
8. Paudyal BP, Gyawalee M, Sigdel K. Eosinophilic Fasciitis: A Rare Fibrosing Disorder. Kathmandu Univ Med J 2012; 39 (3): 73-75.
9. Silny W, Osmola-Mankowska A, Zarnecka-Operacz M, Żaba R, Danczak-Pazdrowska A, Marciniak A. Eosinophilic fasciitis: A report of two cases treated with ultaviolet A1 phototherapy. Photoder Photoimmunol Photomed 2009; 25: 325-327.
10. Lebeaux D, Francès C, Barete S, Wechsler B, Dubourg O, Renoux J y col. Eosinophilic Fasciitis (Shulman Disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology 2012; 51: 557-561.
11. Ackerman AB, Chongchitnant N, Sánchez J y col. Histologic Diagnosis of Inflammatory skin diseases. Segunda Edición. Williams & Wilkins. Philadelphia. EEUU. 1997.